


 Sign Out
Sign Out

Pharmacology: Mechanism of action: Ranibizumab is a humanized recombinant monoclonal antibody fragment targeted against human vascular endothelial growth factor A (VEGF-A). It binds with high affinity to the VEGF-A isoforms (e.g. VEGF110, VEGF121 and VEGF165), thereby preventing binding of VEGF-A to its receptors VEGFR-1 and VEGFR-2.
Pharmacodynamics: Binding of VEGF-A to its receptors leads to endothelial cell proliferation and neovascularization, as well as vascular leakage, which are thought to contribute to the progression of the neovascular form of age-related macular degeneration, to the development of CNV, including CNV secondary to pathologic myopia, or to the macular edema causing visual impairment in diabetes and retinal vein occlusion.
Clinical Studies: Treatment of wet AMD: In wet AMD, the clinical safety and efficacy of ranibizumab have been assessed in three randomized, double-masked, sham** or active-controlled studies in patients with neovascular AMD (FVF2598g (MARINA), FVF2587g (ANCHOR) and FVF3192g (PIER)). A total of 1,323 patients (879 active and 444 control) were enrolled in these studies.
Study FVF2598g (MARINA) and study FVF2587g (ANCHOR): In the 24-month study FVF2598g (MARINA), patients with minimally classic or occult with no classic CNV received monthly intravitreal injections of ranibizumab 0.3 mg or 0.5 mg or sham injections. A total of 716 patients were enrolled in this study (sham, 238; ranibizumab 0.3 mg, 238; ranibizumab 0.5 mg, 240).
In the 24-month study FVF2587g (ANCHOR), patients with predominantly classic CNV lesions received either: 1) monthly intravitreal injections of ranibizumab 0.3 mg and sham PDT; 2) monthly intravitreal injections of ranibizumab 0.5 mg and sham PDT; or 3) sham intravitreal injections and active verteporfin PDT. Verteporfin (or sham) PDT was given with the initial ranibizumab (or sham) injection and every 3 months thereafter if fluorescein angiography showed persistence or recurrence of vascular leakage. A total of 423 patients were enrolled in this study (ranibizumab 0.3 mg, 140; ranibizumab 0.5 mg, 140; verteporfin PDT, 143).
** The sham ranibizumab injection control procedure involved anesthetizing the eye in a manner identical to a ranibizumab intravitreal injection. The tip of a needleless syringe was then pressed against the conjunctiva and the plunger of the needleless syringe depressed.
Key outcomes are summarized in Tables 1, 2 and Figure 1. (See Tables 1, 2 and Figure 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image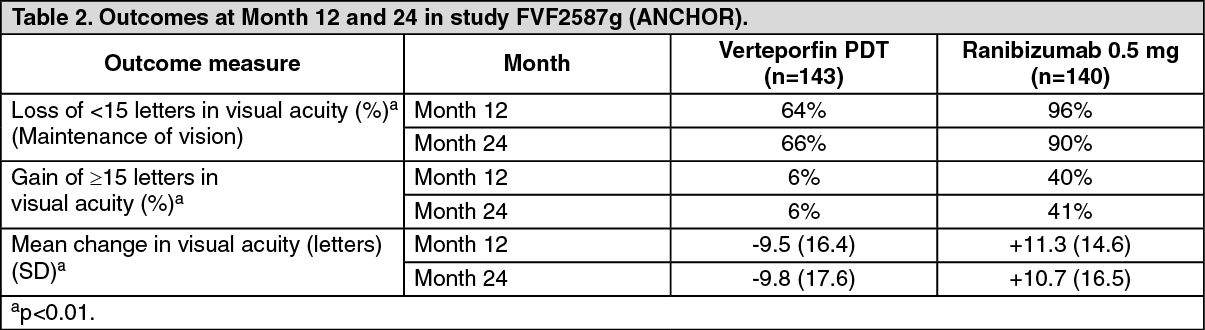 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image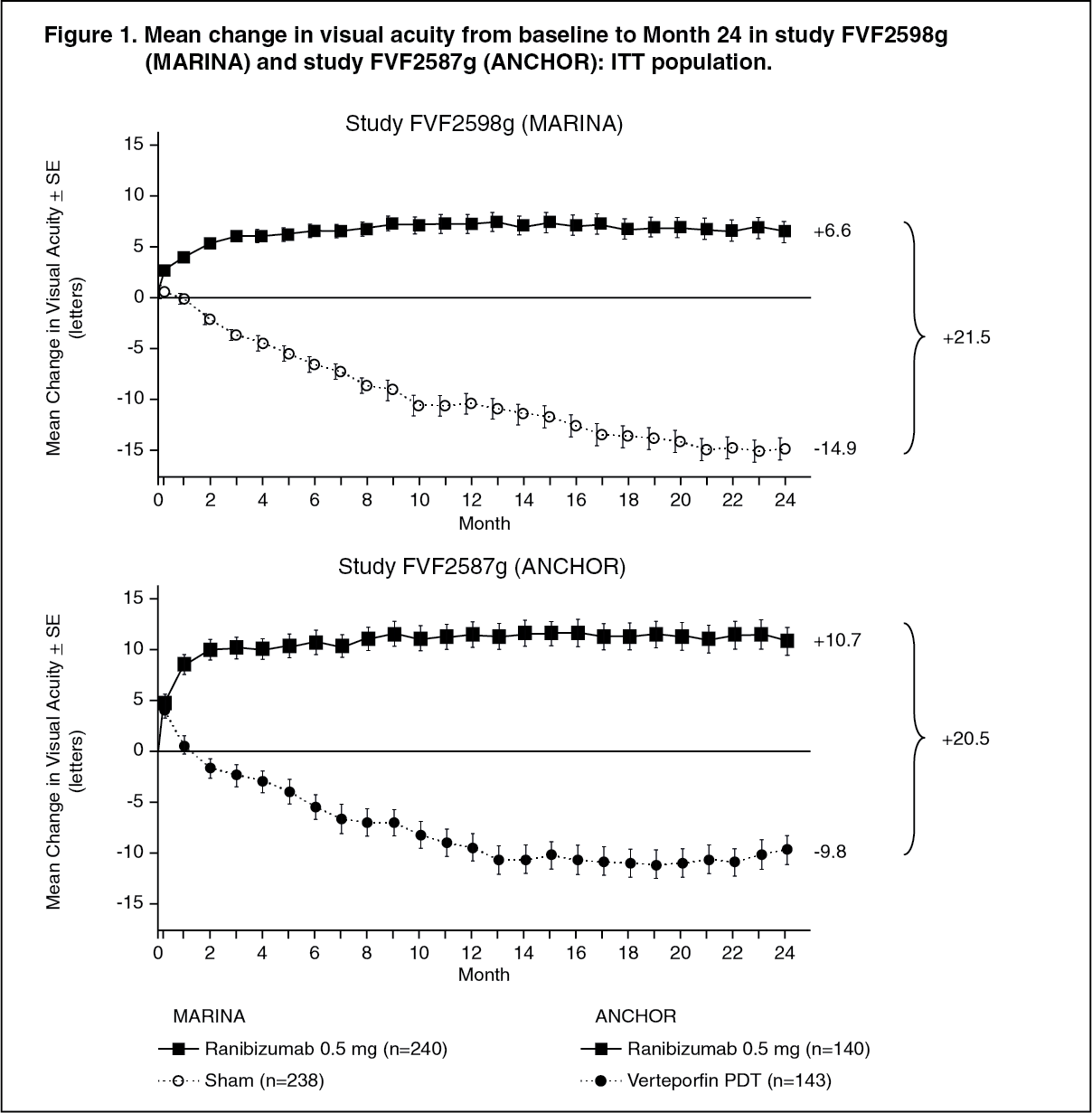 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePatients in the group treated with ranibizumab had minimal observable CNV lesion growth, on average. At Month 12, the mean change in the total area of the CNV lesion was 0.1 to 0.3 DA for ranibizumab versus 2.3 to 2.6 DA for the control arms.
Results from both trials indicated that continued ranibizumab-treatment may be of benefit also in patients who lost ≥15 letters of best-corrected visual acuity (BCVA) in the first year of treatment.
In both the MARINA and ANCHOR studies, the improvement in visual acuity seen with ranibizumab 0.5 mg at 12 months was accompanied by patient-reported benefits as measured by the National Eye Institute Visual Function Questionnaire (VFQ-25) scores. The differences between ranibizumab 0.5 mg and the two control groups were assessed with p-values ranging from 0.009 to <0.0001.
Study FVF3192g (PIER): Study FVF3192g (PIER) was a randomized, double-masked, sham-controlled, two-year study designed to assess the safety and efficacy of ranibizumab in 184 patients with neovascular AMD (with or without a classic CNV component). Patients received ranibizumab 0.3 mg or 0.5 mg intravitreal injections or sham injections once a month for 3 consecutive doses, followed by a dose administered once every 3 months. From Month 14 of the study, sham-treated patients were allowed to cross over to receive ranibizumab and from Month 19, more frequent treatments were possible. Patients treated with ranibizumab in PIER received a mean of 10 treatments during the study. The primary efficacy endpoint was mean change in visual acuity at Month 12 compared with baseline. After an initial increase in visual acuity (following monthly dosing), on average, patients dosed once every three months with ranibizumab lost visual acuity, returning to baseline at Month 12. This effect was maintained in most ranibizumab-treated patients (82%) at Month 24. Data from a limited number of subjects that crossed over to receive ranibizumab after more than a year of sham-treatment suggested that early initiation of treatment may be associated with a better preservation of visual acuity.
Study FVF3689g (SAILOR): Study FVF3689g (SAILOR) was a Phase IIIb, single-masked, one-year multicenter study in naïve and previously treated subjects with CNV secondary to AMD. The primary study objective was to estimate the incidence of ocular and non-ocular serious adverse events in subjects treated for 12 months. Overall, 2378 patients were randomized in a 1:1 ratio to receive one intravitreal injection of 0.3 mg or 0.5 mg ranibizumab every month for three consecutive months followed by re-treatment as needed not more often than monthly.
Overall, no imbalances between the two dose groups were observed in the frequency of ocular and non-ocular adverse events. There was a statistically non-significant trend towards a higher stroke rate in the 0.5 mg group compared to the 0.3 mg group. The respective 95% CIs for the overall stroke rate were wide (0.3% to 1.3% for the 0.3 mg group vs. 0.7% to 2.0% for the 0.5 mg group). The number of strokes was small in both dose groups, and there is not sufficient evidence to conclude (or rule out) that there is a true difference in stroke rates among the treatment groups. The difference in stroke rates may be greater in patients with known risk factors for stroke, including history of prior stroke and transient ischemic attack.
Study A2412 (EVEREST II): Study A2412 (EVEREST II) is a two-year, randomized, double-masked, multicenter study designed to evaluate the efficacy and safety of ranibizumab 0.5 mg monotherapy vs. ranibizumab 0.5 mg in combination with verteporfin photodynamic therapy (vPDT) in 322 Asian patients with symptomatic macular polypoidal choroidal vasculopathy (PCV), a subtype of wet AMD. Patients in both study arms initiated treatment with three monthly ranibizumab injections, plus sham or active vPDT given with the first ranibizumab injection only. Following treatment initiation, ranibizumab monotherapy and ranibizumab administered with vPDT were given pro re nata (PRN) based on ocular clinical assessments, including imaging techniques (e.g. OCT, FA, ICGA). Primary results at Month 12 demonstrated that ranibizumab administered with vPDT was superior to ranibizumab monotherapy with respect to the BCVA change from baseline (8.3 letters versus 5.1 letters, p=0.013) and complete polyp regression (69.3% versus 34.7%, p<0.001). Patients administered ranibizumab with vPDT received on average 2.3 ranibizumab injections less than patients administered ranibizumab monotherapy (5.1 vs. 7.4 injections).
Superiority of ranibizumab with vPDT compared to ranibizumab monotherapy was confirmed at Month 24 with respect to BCVA change from baseline (9.6 letters vs. 5.5 letters, p=0.005) and complete polyp regression (56.6% versus 26.7%, p<0.0001). Patients administered ranibizumab with vPDT received on average 4.2 ranibizumab injections less than patients administered ranibizumab monotherapy (8.1 vs. 12.3 injections).
The safety profile in these patients was consistent with that seen in previous clinical trials with ranibizumab monotherapy.
Treatment of visual impairment due to DME: The efficacy and safety of ranibizumab have been assessed in two randomized, double-masked, sham- or active controlled studies of 12 months duration in patients with visual impairment due to diabetic macular edema (Study D2301 (RESTORE) and D2201 (RESOLVE)). A total of 496 patients (336 active and 160 control) were enrolled in these studies, the majority had type II diabetes, 28 patients treated with ranibizumab had type I diabetes.
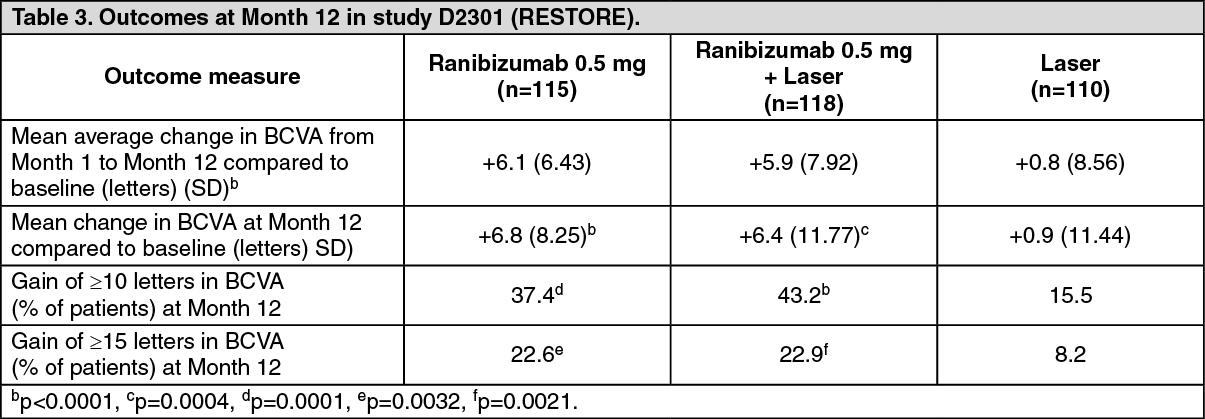
Study D2301 (RESTORE): In study D2301 (RESTORE), a total of 345 patients with visual impairment due to macular edema were randomized to receive either initial intravitreal injections of ranibizumab 0.5 mg as monotherapy and sham laser photocoagulation (n=116), combined ranibizumab 0.5 mg and laser photocoagulation (n=118), or sham** injection and laser photocoagulation (n=111). Treatment with ranibizumab was started with monthly intravitreal injections and continued until visual acuity was stable for at least three consecutive monthly assessments. The treatment was reinitiated when there was a reduction in BCVA due to DME progression. Laser photocoagulation was administered at baseline on the same day, at least 30 minutes before the injection of ranibizumab, and then as needed based on Early Treatment Diabetic Retinopathy Study (ETDRS) criteria.
Key outcomes are summarized in Table 3 and Figure 2. (See Table 3 and Figure 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image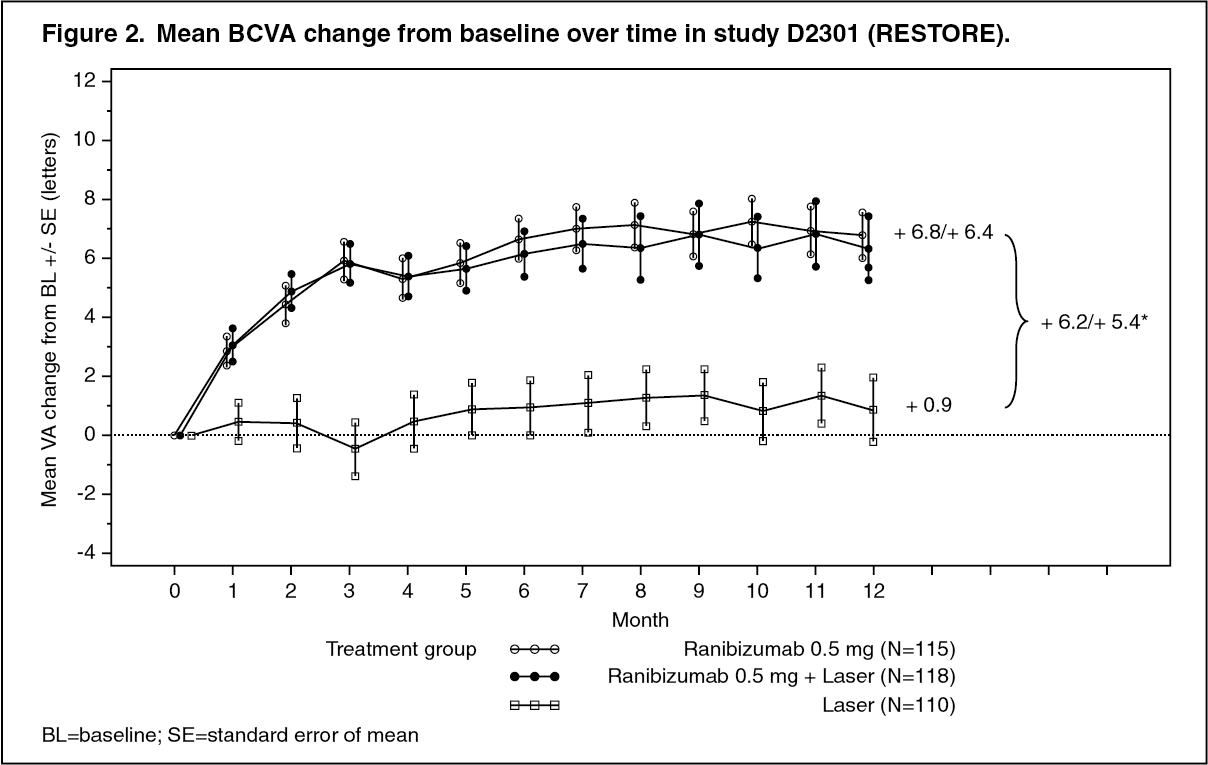 Click on icon to see table/diagram/image
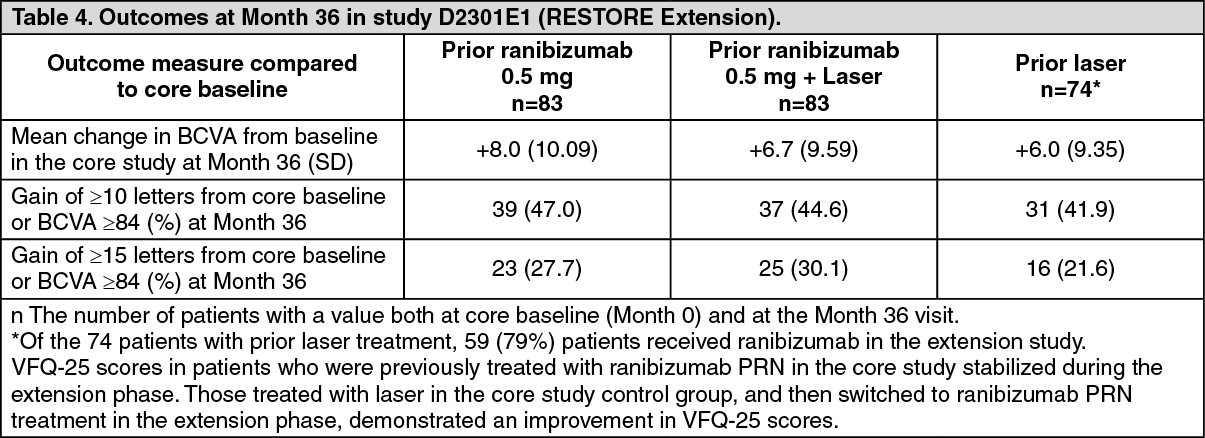
Click on icon to see table/diagram/imageStudy D2301E1 (RESTORE Extension): Study D2301E1 (RESTORE Extension) was an open-label, multi-center, 24-month extension study. 240 patients who had completed the 12-month core study entered the extension study and were treated with ranibizumab 0.5 mg pro re nata (PRN) in the same eye that was selected as the study eye in the core study. Treatment was administered monthly upon a decrease in BCVA due to DME until stable BCVA was reached. In addition, laser treatment was administered, if deemed necessary by the investigator, and based on ETDRS guidelines.
On average, 6.4 ranibizumab injections were administered per patient in the 24-month extension period in patients who were treated with ranibizumab in the core study. Of the 74 patients from the core study laser treatment arm, 59 (79%) patients received ranibizumab at some point during the extension phase. On average, these 59 patients received 8.1 ranibizumab injections per patient over the 24 months of the extension study. The proportions of patients who did not require any ranibizumab treatment during the extension phase were 19%, 25% and 20% in the prior ranibizumab, prior ranibizumab + laser, and prior laser group, respectively.
Key outcome measures are summarized in Table 4. (See Table 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe long-term safety profile of ranibizumab observed in this 24-month extension study is consistent with the known ranibizumab safety profile.
Study D2201 (RESOLVE): In study D2201 (RESOLVE), a total of 151 patients with macular center involvement causing visual impairment were treated with ranibizumab (6 mg/ml, n=51, 10 mg/ml, n=51) or sham (n=49) by monthly intravitreal injections until pre-defined treatment stopping criteria were met. The initial ranibizumab dose (0.3 mg or 0.5 mg) could be doubled at any time during the study after the first injection if the investigator evaluated that response to treatment was not sufficiently achieved. Laser photocoagulation rescue treatment was allowed from Month 3 in both treatment arms. The study was comprised of two parts: an exploratory part (the first 42 patients analyzed at Month 6) and a confirmatory part (the remaining 109 patients analyzed at Month 12).
Key outcomes from the confirmatory part of the study (2/3 of the patients) are summarized in Table 5 and Figure 3. (See Table 5 and Figure 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image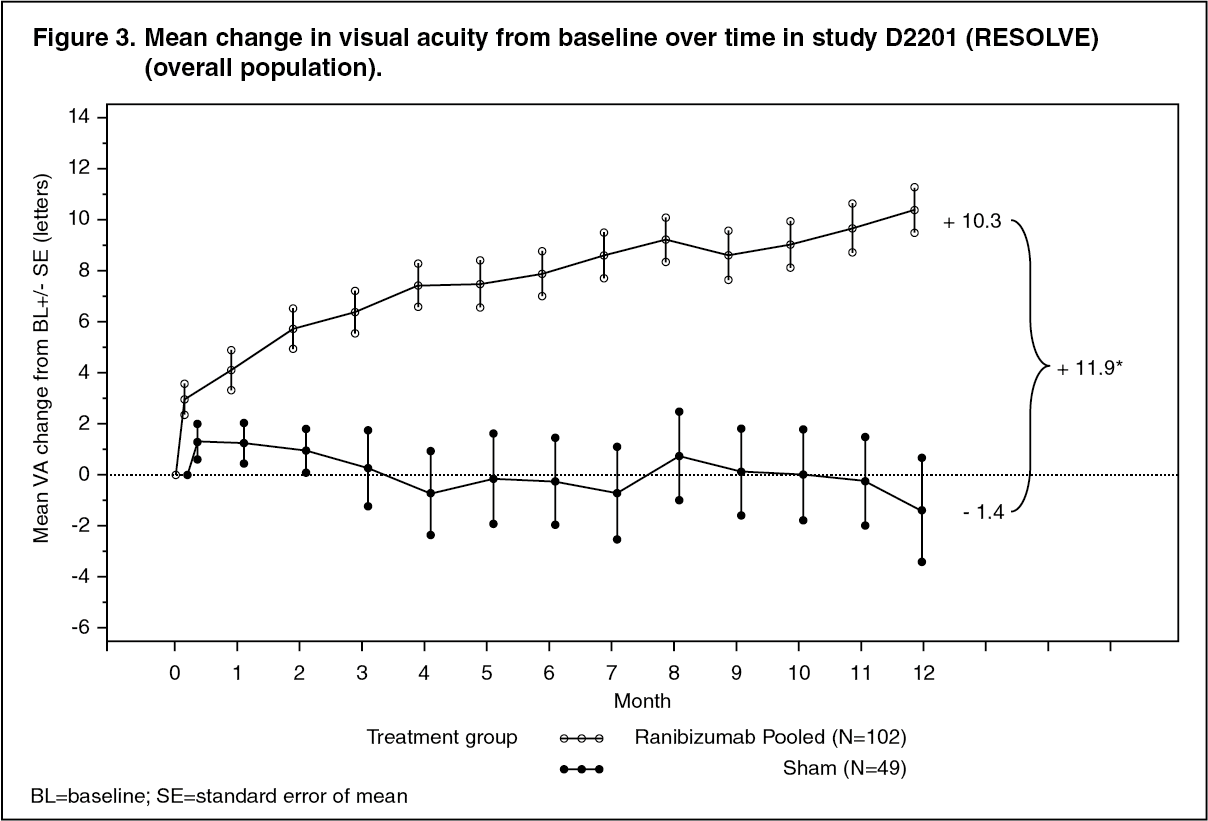 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePatients treated with ranibizumab experienced a continuous reduction in central retina thickness (CRT). At month 12, the mean CRT change from baseline was -194 micrometers for ranibizumab versus -48 micrometers for sham control.
Overall, ocular and non-ocular safety findings in DME patients of both studies D2201 and D2301 were comparable with the previously known safety profile observed in wet AMD patients.
Study D2304 (RETAIN): In the phase IIIb study D2304 (RETAIN), 372 patients with visual impairment due to DME were randomized to receive either intravitreal injection of: ranibizumab 0.5 mg with concomitant laser photocoagulation on a treat-and-extend (TE) regimen (n=121), ranibizumab 0.5 mg monotherapy on a TE regimen (n=128), or ranibizumab 0.5 mg monotherapy on a pro re nata (PRN) regimen (n=123).
In all groups, treatment with ranibizumab was initiated with monthly intravitreal injections and continued until BCVA was stable for at least three consecutive monthly assessments. Laser photocoagulation was administered at baseline on the same day as the first ranibizumab injection and then as needed based on ETDRS criteria. On TE regimen, ranibizumab was then administered, at scheduled treatment at intervals of 2-3 months. On PRN regimen, BCVA was assessed monthly and ranibizumab was administered during the same visit, if needed. In all groups, monthly treatment was re-initiated upon a decrease in BCVA due to DME progression and continued until stable BCVA was reached again. The duration of the study was 24 months.
In the RETAIN study the number of scheduled treatment visits required by the TE regimen was 40% lower than the number of monthly visits required by the PRN regimen. With both regimens, more than 70% of patients were able to maintain their BCVA with a visit frequency of ≥ 2 months.
Key outcome measures are summarized in Table 6. (See Table 6.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn DME studies, the improvement in BCVA was accompanied by a reduction over time in mean CRT in all the treatment groups.
There was no difference in the BCVA or CRT outcomes of patients in RETAIN study who received or did not receive concomitant thiazolidinediones.
Study D2303 (REVEAL): The study D2303 (REVEAL), was a 12 month, randomized, double-masked Phase IIIb trial conducted in Asian patients. Similar to the RESTORE 12 month core study in trial design and inclusion/exclusion criteria, 390 patients with visual impairment due to macular edema were randomized to receive either ranibizumab 0.5 mg injection as monotherapy and sham laser photocoagulation (n=133), ranibizumab 0.5 mg injection and laser photocoagulation (n=129), or sham injection and laser photocoagulation (n=128). Mean change in visual acuity at Month 12 compared to baseline were +6.6 letters in the ranibizumab monotherapy group, +6.4 letters in the ranibizumab plus laser group and +1.8 letters in the laser group. Overall, the efficacy and safety results of the REVEAL study in Asian DME patients are consistent with those of the RESTORE study in Caucasian DME patients.
Diabetic retinopathy severity score (DRSS) was assessed in three of the clinical trials described previously. Of the 875 patients of whom approximately 75% were of Asian origin. In a meta-analysis of these studies, 48.8% of the 315 patients with gradable DRSS scores in the subgroup of patients with moderately severe non-proliferative DR (NPDR) or worse at baseline experienced a ≥2-step improvement in the DRSS at month 12 when treated with ranibizumab (n=192) vs 14.6% of patients treated with laser (n=123). The estimated difference between ranibizumab and laser was 29.9% (95% CI: [20.0, 39.7]). In the 405 DRSS gradable patients with moderate NPDR or better, a ≥2-step DRSS improvement was observed in 1.4% and 0.9% of the ranibizumab and laser groups respectively.
Treatment of PDR: The clinical safety and efficacy of ranibizumab in patients with PDR have been assessed in Protocol S which evaluated the treatment with ranibizumab 0.5 mg intravitreal injections compared with panretinal photocoagulation (PRP). The primary endpoint was the mean visual acuity change at year 2. Additionally, change in diabetic retinopathy (DR) severity was assessed based on fundus photographs using the DR severity score (DRSS).
Protocol S was a multicentre, randomized, active-controlled, parallel-assignment, non-inferiority phase III study in which 305 patients (394 study eyes) with PDR with or without DME at baseline were enrolled. The study compared ranibizumab 0.5 mg intravitreal injections to standard treatment with PRP. A total of 191 eyes (48.5%) were randomized to ranibizumab 0.5 mg and 203 eyes (51.5%) eyes were randomized to PRP. A total of 88 eyes (22.3%) had baseline DME: 42 (22.0%) and 46 (22.7%) eyes in the ranibizumab and PRP groups, respectively.
In this study, the baseline visual acuity was 75.0 letters in the ranibizumab group and 75.2 letters in the PRP group, the mean visual acuity change at year 2 was +2.7 letters in the ranibizumab group compared to -0.7 letters in the PRP group. The difference in least square means was 3.5 letters (95% CI: [0.2 to 6.7]).
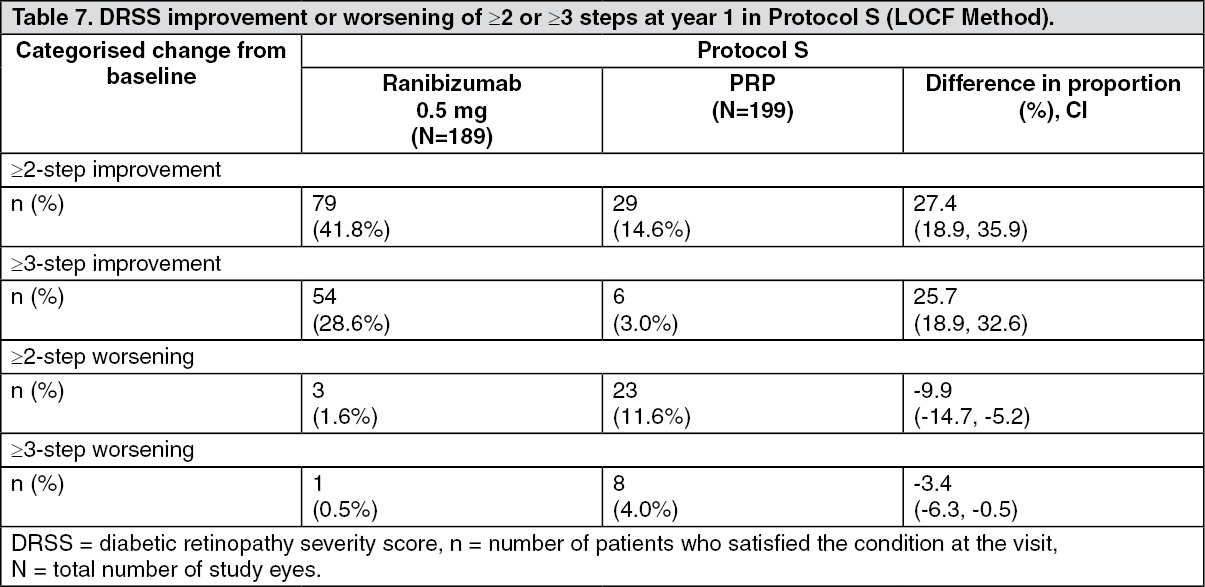
At year 1, 41.8% of eyes experienced a ≥2-step improvement in the DRSS when treated with ranibizumab (n=189) compared to 14.6% of eyes treated with PRP (n=199). The estimated difference between ranibizumab and laser was 27.4% (95% CI: [18.9, 35.9]). (See Table 7.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAt year 1 in the ranibizumab-treated group in Protocol S, ≥2-step improvement in DRSS was consistent in eyes without DME (39.9%) and with baseline DME (48.8%).
An analysis of year 2 data from Protocol S demonstrated that 42.3% (n=80) of eyes in the ranibizumab-treated group had ≥2-step improvement in DRSS from baseline compared with 23.1% (n=46) of eyes in the PRP group. In the ranibizumab-treated group, ≥2-step improvement in DRSS from baseline was observed in 58.5% (n=24) of eyes with baseline DME and 37.8% (n=56) of eyes without DME.
Treatment of visual impairment due to macular edema secondary to RVO: Study FVF4165g (BRAVO) and study FVF4166g (CRUISE): The clinical safety and efficacy of ranibizumab in patients with visual impairment due to macular edema secondary to RVO have been assessed in the randomized, double-masked, controlled studies BRAVO and CRUISE that recruited subjects with BRVO (n=397) and CRVO (n=392), respectively. In both studies, subjects received either 0.3 mg or 0.5 mg intravitreal ranibizumab or sham** injections. After 6 months, patients in the sham-control arms were crossed over to 0.5 mg ranibizumab. In BRAVO, laser photocoagulation as rescue was allowed in all arms from Month 3.
Key outcomes from BRAVO and CRUISE are summarized in Tables 8 and 9, and Figures 4 and 5. (See Tables 8 and 9, and Figures 4 and 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image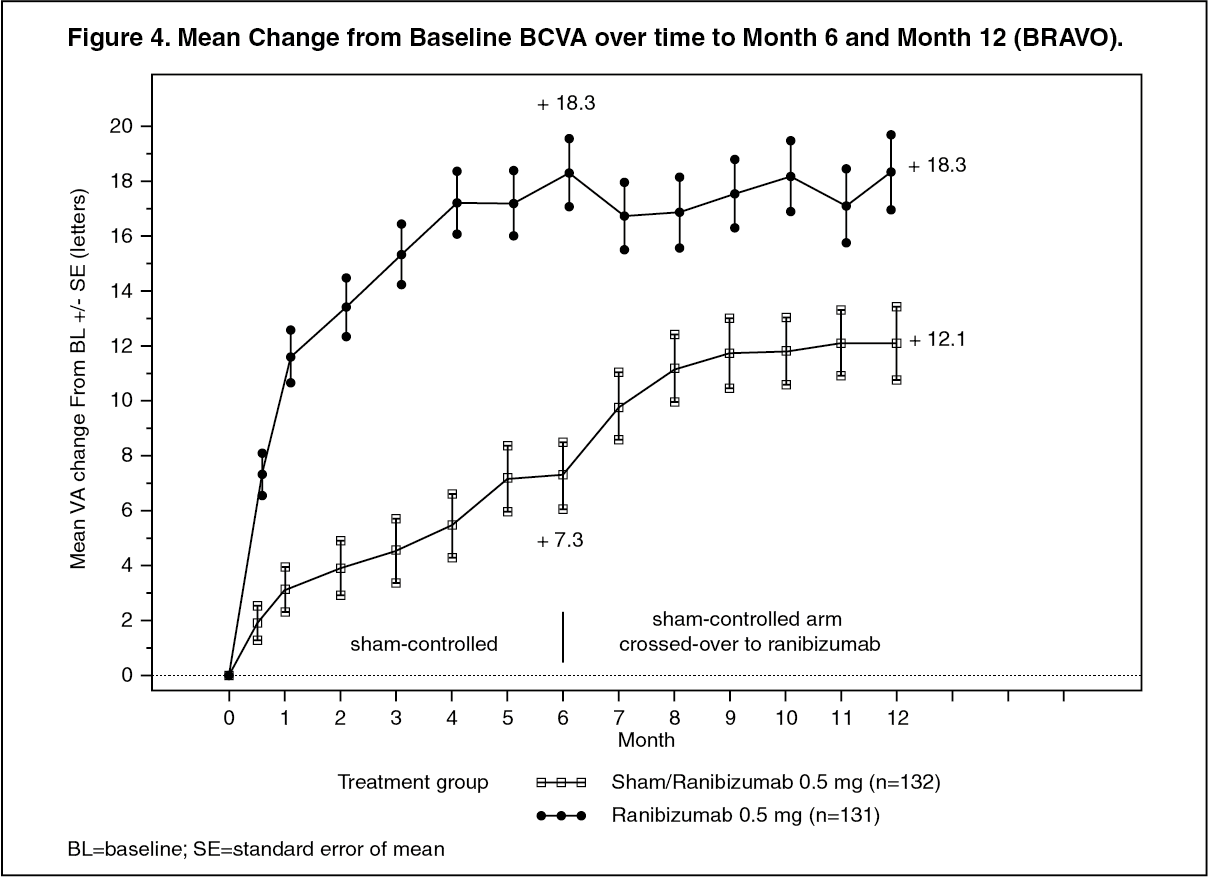 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image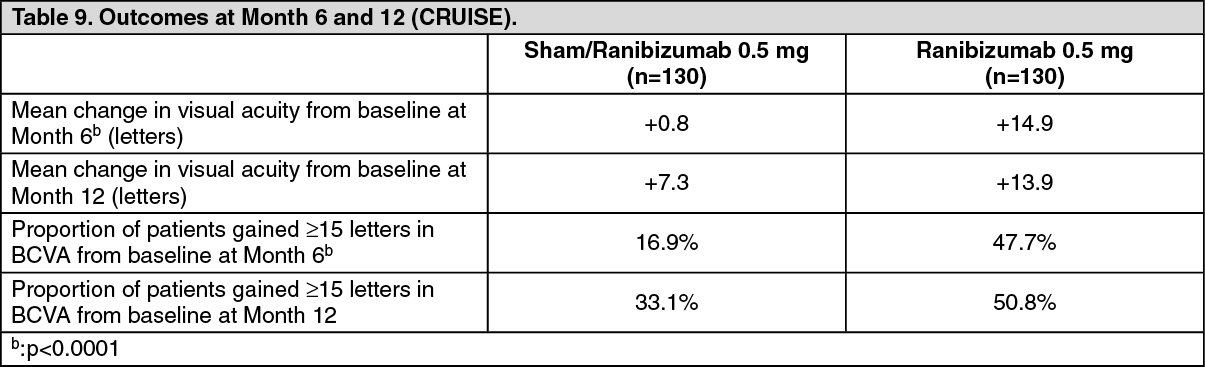 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image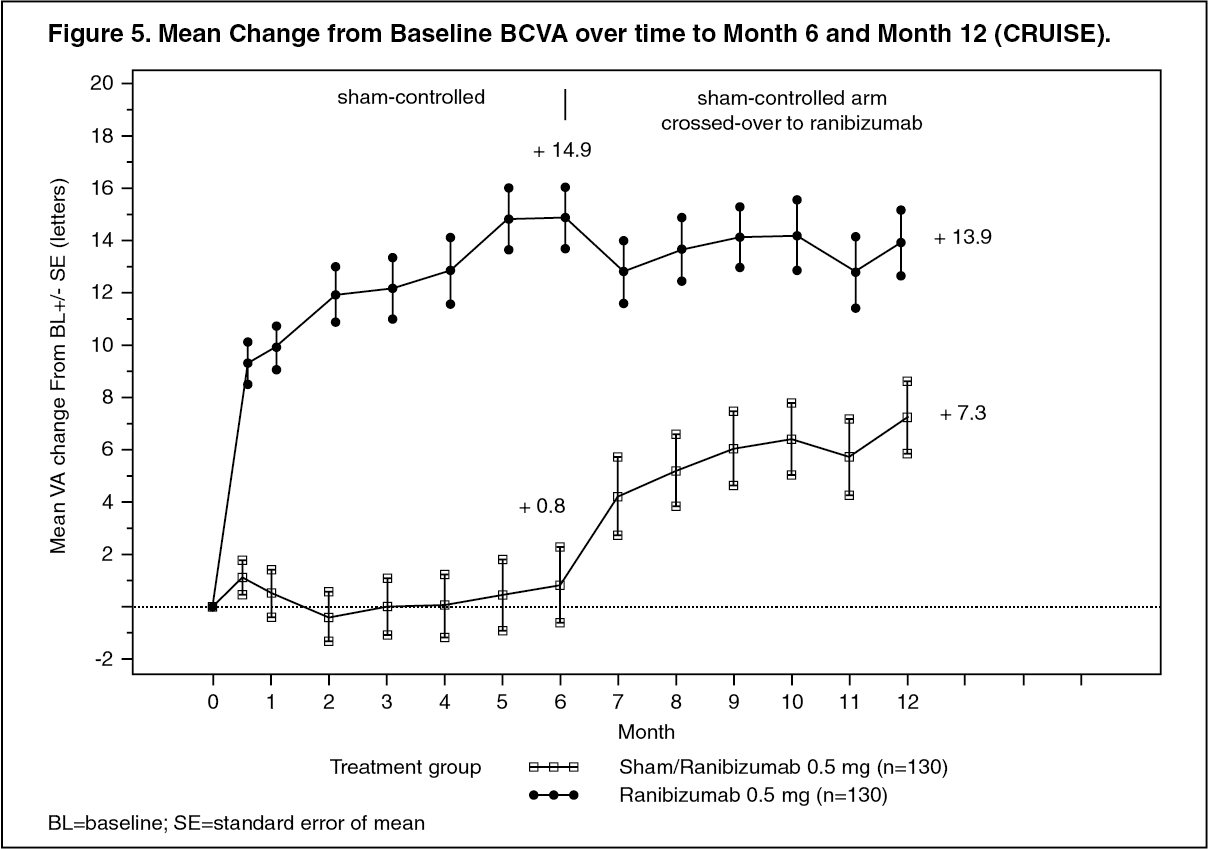 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn both studies, the improvement of vision was accompanied by a continuous decrease in the macular edema as measured by central retinal thickness.
The improvement in visual acuity seen with ranibizumab treatment at 6 and 12 months was accompanied by patient-reported benefits as measured by the National Eye Institute Visual Function Questionnaire (VFQ-25) sub-scales related to near and distance activity, a pre-specified secondary efficacy endpoint. The difference between ranibizumab 0.5 mg and the control group was assessed at Month 6 with p-values of 0.02 to 0.0002.
Study E2401 (CRYSTAL) and study E2402 (BRIGHTER): The long term (24 month) clinical safety and efficacy of ranibizumab in patients with visual impairment due to macular edema secondary to RVO were assessed in the BRIGHTER and CRYSTAL studies, which recruited subjects with BRVO (n=455) and CRVO (n=357), respectively. In both studies, subjects received a 0.5 mg ranibizumab PRN dosing regimen driven by individualized stabilization criteria. BRIGHTER was a 3-arm, randomized, active-controlled study that compared 0.5 mg ranibizumab given as monotherapy or in combination with adjunctive laser photocoagulation, to laser photocoagulation alone. After 6 months, subjects in the laser monotherapy arm could receive 0.5 mg ranibizumab. CRYSTAL was a single-arm study with 0.5 mg ranibizumab monotherapy.
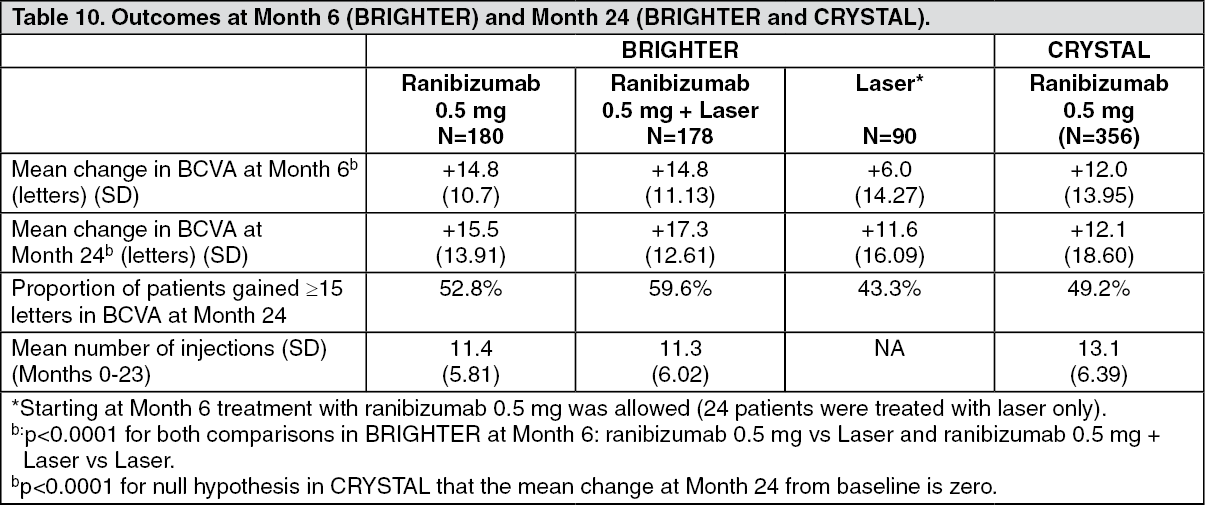
Key outcome measures from BRIGHTER and CRYSTAL are shown in Table 10 and Figures 6 and 7. (See Table 10 and Figures 6 and 7.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image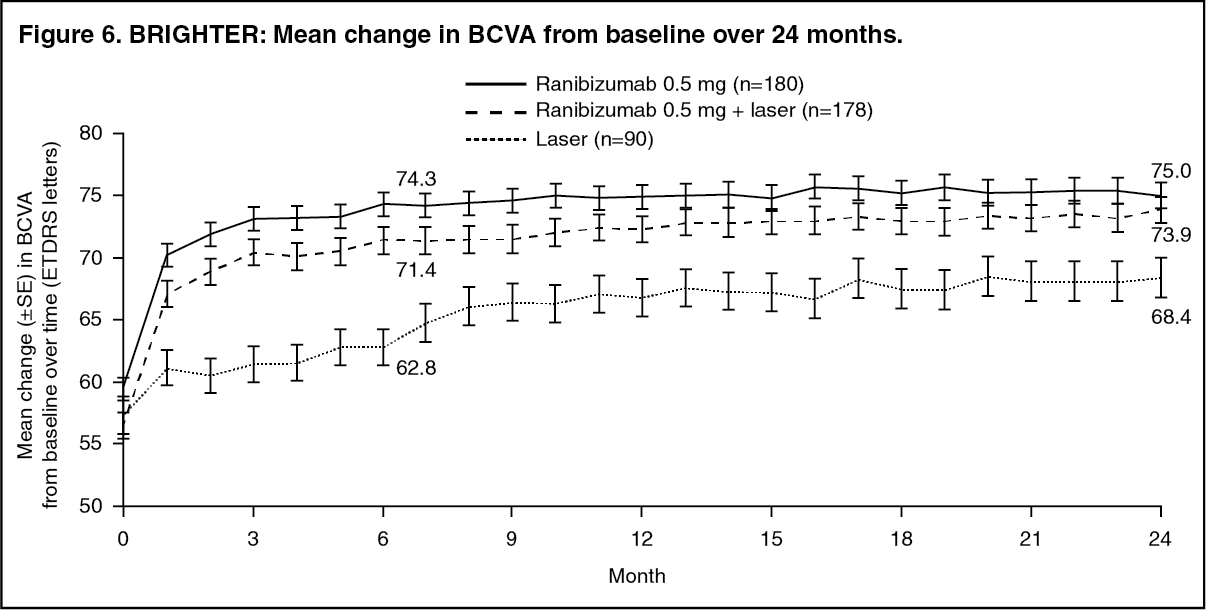 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn BRIGHTER, 0.5 mg ranibizumab with adjunctive laser therapy demonstrated non-inferiority to ranibizumab monotherapy from baseline to Month 24 as assessed by the mean average change in BCVA. There was no difference between the two groups in the number of ranibizumab injections administered over this period.
In both studies, a rapid and significant decrease from baseline in central retinal subfield thickness was observed at Month 1. This effect was maintained up to Month 24.
The beneficial effect of ranibizumab treatment was similar irrespective of the presence of retinal ischemia. In BRIGHTER, patients with retinal ischemia present (N=87) or absent (N=35) and treated with ranibizumab monotherapy had a mean change from baseline of +15.4 and +12.9 letters respectively, at Month 24. In CRYSTAL, patients with retinal ischemia present (N=107) or absent (N=109), treated with ranibizumab monotherapy had a mean change from baseline of +11.1 and +12.9 letters, respectively.
The beneficial effect in terms of visual improvement was observed in all patients treated with 0.5 mg ranibizumab monotherapy regardless of their disease duration in both BRIGHTER and CRYSTAL. In patients with <3 months disease duration an increase in visual acuity of 13.3 and 10.0 letters was seen at Month 1; and 17.7 and 13.2 letters at Month 24 in BRIGHTER and CRYSTAL, respectively. Treatment initiation at the time of diagnosis should be considered.
The long term safety profile of ranibizumab observed in these 24-month studies is consistent with the known ranibizumab safety profile.
Treatment of visual impairment due to CNV: Study G2301 (MINERVA): The clinical safety and efficacy of ranibizumab in patients with visual impairment due to CNV secondary to etiologies other than nAMD and PM have been assessed based on the 12-month data of the randomized, double-masked, sham controlled pivotal study G2301 (MINERVA). Due to the multiple baseline etiologies involved, five subgroups (angioid streaks, post-inflammatory retinochoroidopathy, central serous chorioretinopathy, idiopathic chorioretinopathy, and miscellaneous etiology) were pre-defined for analysis. In this study, 178 patients were randomized in a 2:1 ratio to one of the following arms: ranibizumab 0.5 mg at baseline followed by an individualized dosing regimen driven by disease activity; sham injection at baseline followed by an individualized treatment regimen driven by disease activity.
Starting at Month 2, all patients received open-label treatment with ranibizumab as needed. The primary endpoint was assessed by the best corrected visual acuity (BCVA) change from baseline to Month 2.
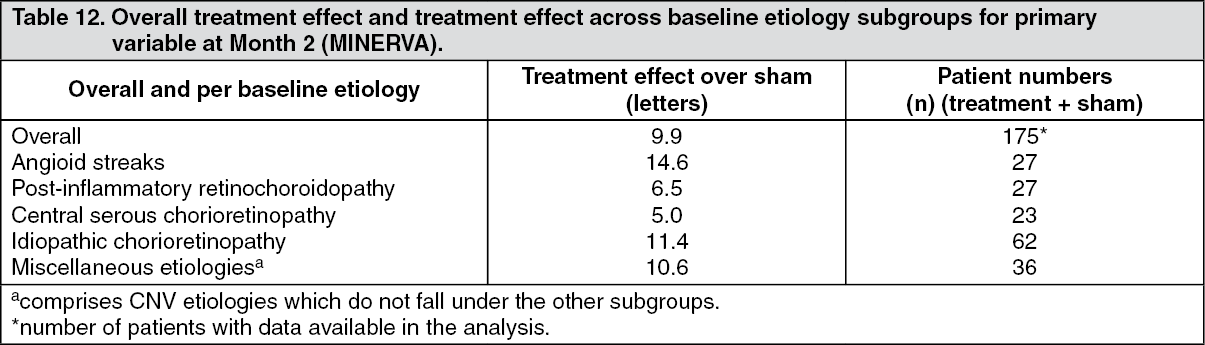
Key outcomes from MINERVA are summarized in Tables 11 and 12 and Figure 8. (See Table 11 and Figure 8.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image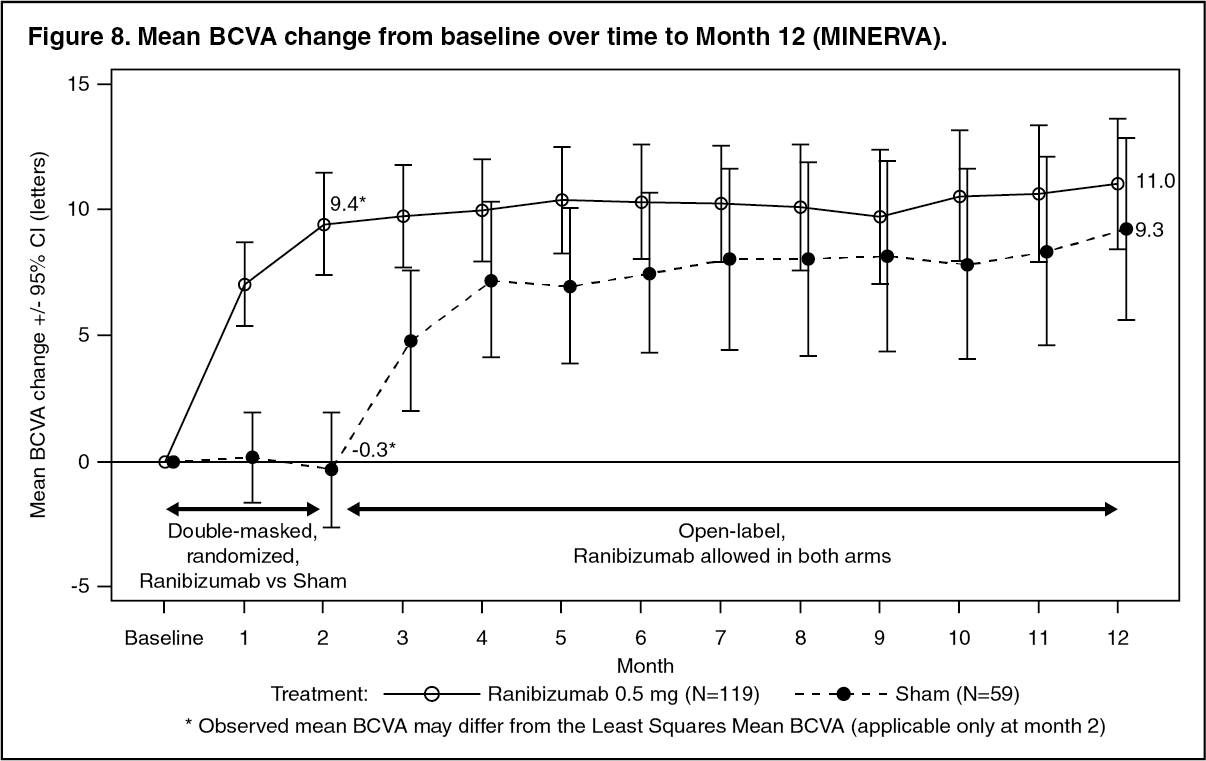 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageWhen comparing ranibizumab versus sham control at Month 2, a consistent treatment effect both overall and across baseline etiology subgroups was observed. (See Table 12.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe improvement of vision was accompanied by a reduction in central subfield thickness over the 12-month period.
The mean number of ranibizumab injections given in the study eye over 12 months was 5.8 in the ranibizumab arm versus 5.4 in those patients in the sham with ranibizumab group. In the sham arm, 7 out of 59 patients did not receive any treatment with ranibizumab in the study eye during the 12-month period.
A trend in patient-reported benefits, as measured by the NEI VFQ-25 composite score, was observed from baseline to Month 2 for patients receiving ranibizumab treatment versus the sham control group. This trend was maintained to Month 12.
Pediatric patients: Five adolescent patients aged 12 to 17 years with visual impairment secondary to CNV received open-label treatment with ranibizumab 0.5 mg at baseline followed by an individualized treatment regimen based on evidence of disease activity (e.g. VA impairment, intra/sub-retinal fluid, hemorrhage or leakage). BCVA change from baseline to Month 12 improved in all five patients, ranging from +5 to +38 letters (mean of 16.6 letters). The improvement of vision was accompanied by a stabilization or reduction in central subfield thickness over the 12-month period. The mean number of ranibizumab injections given in the study eye over 12 months was three (see Special populations: Pediatric population under DOSAGE & ADMINISTRATION).
Treatment of visual impairment due to CNV secondary to PM: Study F2301 (RADIANCE): The clinical safety and efficacy of ranibizumab in patients with visual impairment due to CNV in PM have been assessed based on the 12-month data of the randomized, double-masked, controlled pivotal study F2301 (RADIANCE) which was designed to evaluate two different dosing regimens of 0.5 mg ranibizumab given as intravitreal injection in comparison to verteporfin PDT (vPDT, Visudyne photodynamic therapy).
The 277 patients were randomized to one of the following arms: Group I (ranibizumab 0.5mg, dosing regimen driven by "stability" criteria defined as no change in BCVA compared to two preceding monthly evaluations).
Group II (ranibizumab 0.5mg, dosing regimen driven by "disease activity" criteria defined as vision impairment attributable to intra-or-subretinal fluid or active leakage due to the CNV lesion as assessed by OCT and/or FA).
Group III (vPDT - patients were allowed to receive ranibizumab treatment as of Month 3).
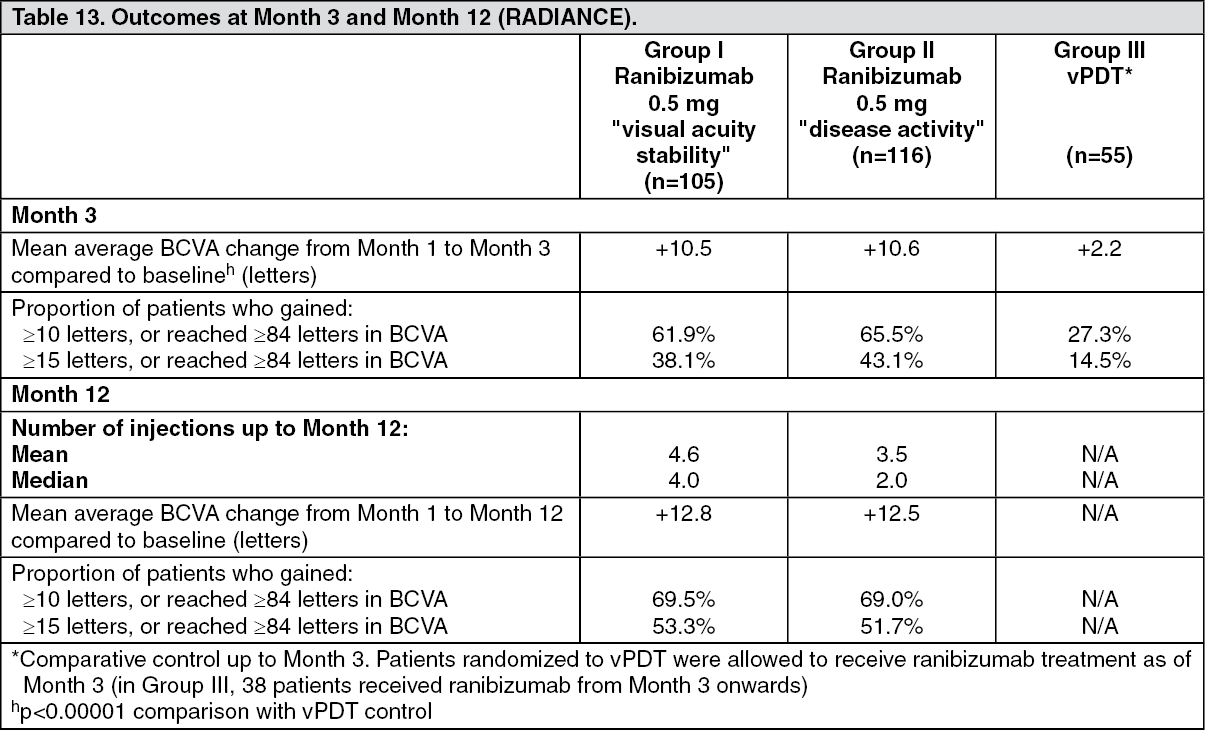
Over the 12 months of the study patients received on average 4.6 injections (range 1-11) in Group I and 3.5 injections (range 1-12) in Group II. In Group II (in which patients received the recommended treatment regimen based on disease activity, see DOSAGE & ADMINISTRATION), 50.9% of patients required 1 or 2 injections, 34.5% required 3 to 5 injections and 14.7% required 6 to 12 injections over the 12-month study period. In Group II, 62.9% of patients did not require injections in the second 6 months of the study.
Key outcomes from RADIANCE are summarized in Table 13 and Figure 9. (See Table 13 and Figure 9.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image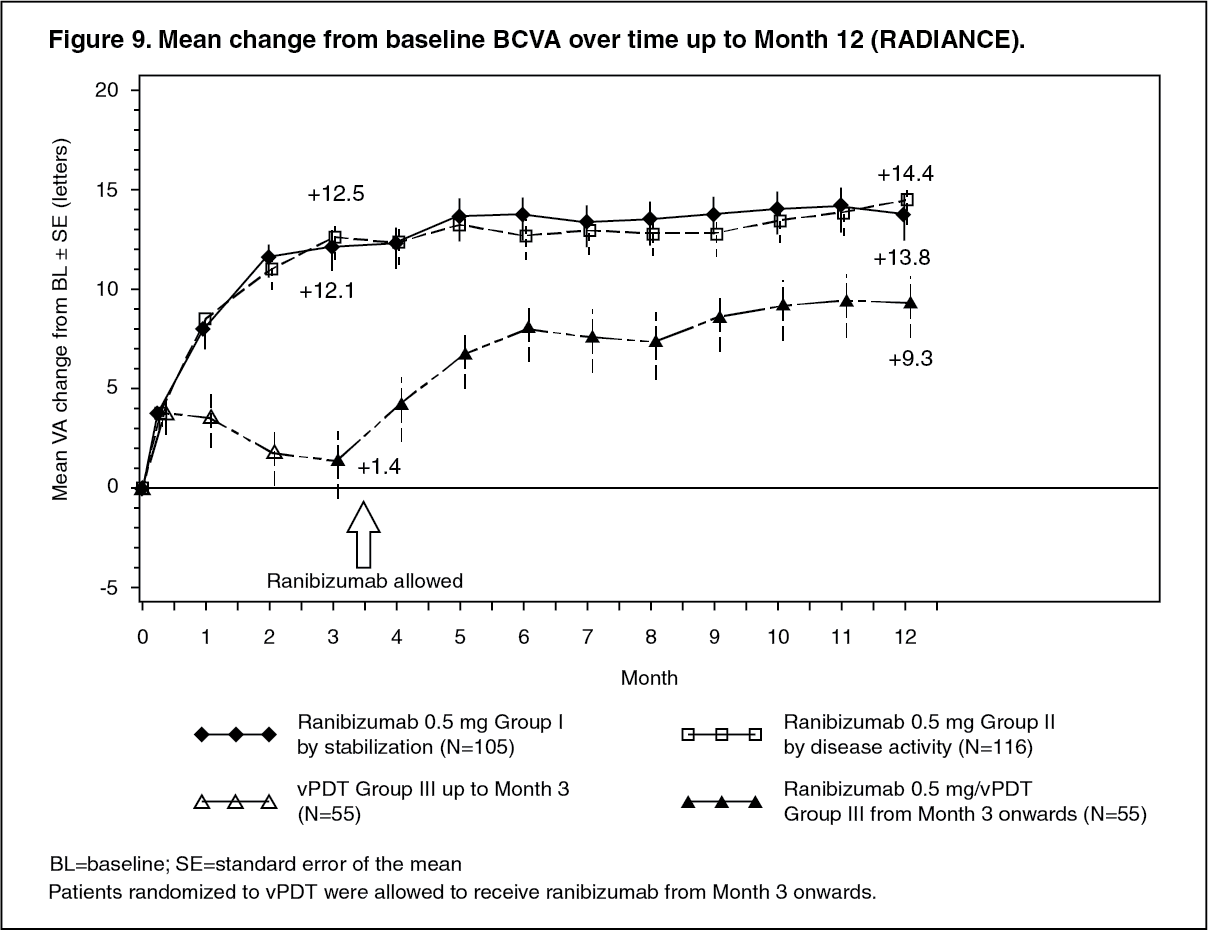 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe improvement of vision was accompanied by a reduction in central retinal thickness.
Patient-reported benefits were observed with the ranibizumab treatment arms over vPDT (p-value <0.05) in terms of improvement in the composite score and several subscales (general vision, near activities, mental health and dependency) of the VFQ-25.
Treatment of ROP in preterm infants: Study H2301 (RAINBOW): The clinical safety and efficacy of Accentrix 0.2 mg for the treatment of ROP in preterm infants have been assessed based on the 6-month data of the randomized, open-label, 3-arm, parallel group, superiority study H2301 (RAINBOW), which was designed to evaluate ranibizumab 0.2 mg and 0.1 mg given as intravitreal injections in comparison to laser therapy. Eligible patients had to have one of the following retinal findings in each eye: Zone I, stage 1+, 2+, 3 or 3+ disease, or; Zone II, stage 3+ disease, or; Aggressive posterior (AP)-ROP.
In this study, 225 patients were randomized in a 1:1:1 ratio to receive intravitreal ranibizumab 0.2 mg (n=74), 0.1 mg (n=77), or laser therapy (n=74).
Treatment success, as measured by the absence of active ROP and absence of unfavorable structural outcomes in both eyes 24 weeks after the first study treatment, was highest with ranibizumab 0.2 mg (80.0%) compared to laser therapy (66.2%). The majority of patients treated with ranibizumab 0.2 mg (78.1%) did not require re-treatment with ranibizumab. The difference between ranibizumab 0.2 mg and laser was clinically relevant with an odds ratio (OR) of 2.19 (95% confidence interval (CI) [0.9932, 4.8235]). The primary endpoint did not reach statistical significance (see Table 14).
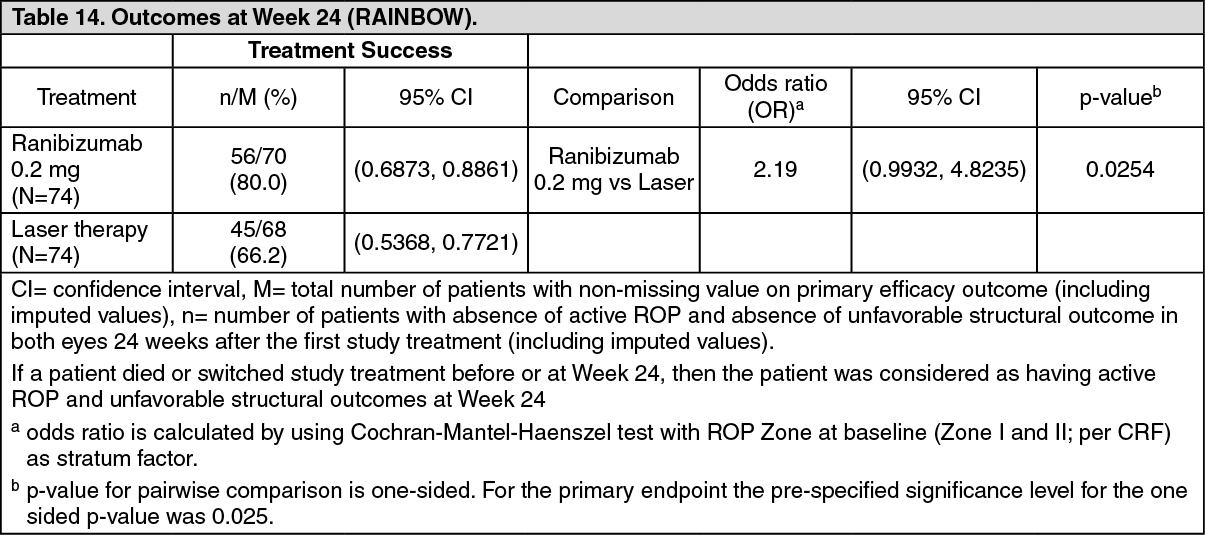 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageFewer patients in the ranibizumab 0.2 mg group switched to another treatment modality due to lack of response compared with the laser group (14.9% vs. 24.3%). Unfavorable structural outcomes were less frequently reported for ranibizumab 0.2 mg (1 patient, 1.4%) compared with laser therapy (7 patients, 10.1%). In addition, 75% of patients achieved resolution of plus disease within 8 days with ranibizumab 0.2 mg compared to 22.5 days in patients treated with laser.
Pharmacokinetics: Absorption: Following monthly intravitreal administration of ranibizumab to patients with neovascular AMD, serum concentrations of ranibizumab were generally low, with maximum levels (Cmax) generally below the ranibizumab concentration necessary to inhibit the biological activity of VEGF by 50% (11 to 27 ng/mL, as assessed in an in vitro cellular proliferation assay). Cmax was dose proportional over the dose range of 0.05 to 1.0 mg/eye. Upon monthly intravitreal administration of ranibizumab 0.5 mg/eye, serum ranibizumab Cmax, attained approximately 1 day after dosing, is predicted to generally range between 0.79 and 2.90 ng/mL, and Cmin is predicted to generally range between 0.07 and 0.49 ng/mL. Serum ranibizumab concentrations in DME and RVO patients were similar to those observed in neovascular AMD patients.
Distribution and elimination: Based on analysis of population pharmacokinetics and the disappearance of ranibizumab from serum for patients with neovascular AMD treated with the 0.5 mg dose, the average vitreous elimination half-life of ranibizumab is approximately 9 days. Serum ranibizumab exposure is predicted to be approximately 90,000-fold lower than vitreal ranibizumab exposure.
Special populations: Pediatric Population (preterm infants with ROP): Following intravitreal administration of ranibizumab to preterm infants with ROP at a dose of 0.2 mg (per eye), serum ranibizumab concentrations were higher than those observed in neovascular AMD adult patients receiving 0.5 mg in one eye. Based on a population pharmacokinetic analysis, the differences in Cmax and AUCinf were approximately 16-fold and 12-fold higher, respectively. The apparent systemic half-life was approximately 6 days. In this analysis, there was no relationship determined between systemic ranibizumab concentrations and systemic VEGF concentrations.
Renal impairment: No formal studies have been conducted to examine the pharmacokinetics of ranibizumab in patients with renal impairment. In a population pharmacokinetic analysis of neovascular AMD patients, 68% (136 of 200) had renal impairment (46.5% mild [50 to 80 mL/min], 20% moderate [30 to 50 mL/min] and 1.5% severe [<30 mL/min]). In RVO patients, 48.2% (253 of 525) had renal impairment (36.4% mild, 9.5% moderate and 2.3% severe). Systemic clearance was slightly lower, but this was not clinically significant.
Hepatic impairment: No formal studies have been conducted to examine the pharmacokinetics of ranibizumab in patients with hepatic impairment.
Toxicology: Non-Clinical Safety Data: Bilateral intravitreal administration of ranibizumab to cynomolgus monkeys at doses between 0.25 mg/eye and 2.0 mg/eye once every 2 weeks for up to 26 weeks resulted in dose-dependent ocular effects.
Intraocularly, there were dose-dependent increases in anterior chamber flare and cells with a peak 2 days after injection. The severity of the inflammatory response generally diminished with subsequent injections or during recovery. In the posterior segment, there were vitreal cell infiltration and floaters, which also tended to be dose-dependent and generally persisted to the end of the treatment period. In the 26-week study, the severity of the vitreous inflammation increased with the number of injections. However, evidence of reversibility was observed after recovery. The nature and timing of the posterior segment inflammation is suggestive of an immune-mediated antibody response, which may be clinically irrelevant. Cataract formation was observed in some animals after a relatively long period of intense inflammation, suggesting that the lens changes were secondary to severe inflammation. A transient increase in post-dose intraocular pressure was observed following intravitreal injections, irrespective of dose.
Microscopic ocular changes were related to inflammation and did not indicate degenerative processes. Granulomatous inflammatory changes were noted in the optic disc of some eyes. These posterior segment changes diminished, and in some instances resolved, during the recovery period.
Following intravitreal administration, no signs of systemic toxicity were detected. Serum and vitreous antibodies to ranibizumab were found in a subset of treated animals.
No carcinogenicity or mutagenicity data are available.
In pregnant monkeys, intravitreal ranibizumab treatment resulting in maximal systemic exposures 0.9-7-fold a worst case clinical exposure did not elicit developmental toxicity or teratogenicity, and had no effect on weight or structure of the placenta, although, based on its pharmacological effect ranibizumab should be regarded as potentially teratogenic and embryo-foetotoxic.
The absence of ranibizumab-mediated effects on embryo-foetal development is plausibly related mainly to the inability of the Fab fragment to cross the placenta. Nevertheless, a case was described with high maternal ranibizumab serum levels and presence of ranibizumab in foetal serum, suggesting that the anti-ranibizumab antibody acted as (Fc region containing) carrier protein for ranibizumab, thereby decreasing its maternal serum clearance and enabling its placental transfer. As the embryo-foetal development investigations were performed in healthy pregnant animals and disease (such as diabetes) may modify the permeability of the placenta towards a Fab fragment, the study should be interpreted with caution.