


 Sign Out
Sign Out

Serious adverse events related to the injection procedure included endophthalmitis, rhegmatogenous retinal detachment, retinal tear and iatrogenic traumatic cataract (see PRECAUTIONS).
Other serious ocular events observed among ranibizumab-treated patients included intraocular inflammation and increased intraocular pressure (see PRECAUTIONS).
The adverse events listed as follows in Table 15 occurred at a higher rate (at least 2 percentage points) in patients receiving treatment with ranibizumab 0.5 mg than in those receiving control treatment (sham injection, as defined in PHARMACOLOGY: PHARMACODYNAMICS under Actions or verteporfin photodynamic therapy (PDT)) in the pooled data of the three controlled wet AMD studies. These were therefore considered potential adverse drug reactions. The safety data described as follows also include all adverse events suspected to be at least potentially related to the injection procedure or medicinal product in the 440 wAMD patients treated with 0.5 mg ranibizumab.
DME population: The safety of ranibizumab was studied in a one-year sham-controlled trial (RESOLVE) and in a one-year laser-controlled trial (RESTORE) conducted respectively in 102 and 235 ranibizumab-treated patients with visual impairment due to DME (see Pharmacology: Pharmacodynamics: CLINICAL STUDIES under Actions). The event of urinary tract infection, in the common frequency category, met the adverse reaction criteria for the Table 15 as follows; otherwise ocular and non-ocular events in the RESOLVE and RESTORE trials were reported with a frequency and severity similar to those seen in the wet AMD trials.
Post-Registration Study in DME population: An analysis of 24-month data from two Phase III studies in DME, RIDE and RISE, is available. Both studies are randomised, sham-controlled studies of monthly intravitreal ranibizumab injections (0.5 mg or 0.3 mg) for a total of 36 months in patients with clinically significant macular oedema with centre involvement secondary to diabetes mellitus (type 1 or type 2). The patients are treated using a fixed dosing regimen which requires monthly injections as opposed to the approved individualised dosing regimen (see DOSAGE & ADMINISTRATION). A total of 500 patients were exposed to ranibizumab treatment in the pooled studies (250 patients in each pooled ranibizumab 0.3mg and 0.5mg arm as well as the sham arm.
The pooled safety analysis showed a numerically higher, but not statistically significant, number of deaths and cerebrovascular events in the 0.5mg group as compared to the 0.3mg or sham groups. The stroke rate at 2 years was 3.2% (8/250) with ranibizumab 0.5mg, 1.2% (3/250) with ranibizumab 0.3mg, and 1.6% (4/250) with sham. Fatalities in the first 2 years occurred in 4.4% (11/250) of patients treated with ranibizumab 0.5mg, in 2.8% (7/250) treated with ranibizumab 0.3mg, and in 1.2% (3/250) of control patients.
PDR population: The safety of ranibizumab in patients with PDR was studied for up to 24-months in Protocol S, including 191 patients treated with ranibizumab 0.5 mg (see Pharmacology: Pharmacodynamics: CLINICAL STUDIES under Actions). Ocular and non-ocular events observed were consistent with what would be expected in a diabetic patient population with DR, or have been reported with a frequency and severity similar to those seen in previous clinical trials with ranibizumab.
RVO population: The safety of ranibizumab was studied in two 12-month trials (BRAVO and CRUISE) conducted respectively in 264 and 261 ranibizumab-treated patients with visual impairment due to macular edema secondary to BRVO and CRVO, respectively (see Pharmacology: Pharmacodynamics: CLINICAL STUDIES under Actions). Ocular and non-ocular events in the BRAVO and CRUISE trials were reported with a frequency and severity similar to those seen in the wet AMD trials.
CNV population: The safety of ranibizumab was studied in a 12-month clinical trial (MINERVA), which included 171 ranibizumab-treated patients with visual impairment due to CNV (see Pharmacology: Pharmacodynamics: CLINICAL STUDIES under Actions). The safety profile in these patients was consistent with that seen in previous clinical trials with ranibizumab.
Pathologic Myopia (PM) population: The safety of ranibizumab was studied in the 12-month clinical trial (RADIANCE), which included 224 ranibizumab-treated patients with visual impairment due to CNV secondary to PM (see Pharmacology: Pharmacodynamics: CLINICAL STUDIES under Actions). Ocular and non-ocular events in this trial were reported with a frequency and severity similar to those seen in the wet AMD trials.
Patients with PM have an increased risk for retinal detachment and retinal tear. No case of 'retinal detachment' was reported in the pivotal clinical trial (RADIANCE) in PM and three events coded as 'retinal tear' were reported. This incidence (1.3%) is higher than that seen in other approved indications for ranibizumab (0 to 1.1% in wet AMD, 0 to 0.8% in DME and in RVO) and consistent with the reporting rate for retinal tear described in Table 15.
Tabulated summary of adverse drug reactions from clinical trials: The adverse drug reactions from clinical trials (Table 15) are listed by MedDRA system organ class. Within each system organ class, the adverse drug reactions are ranked by frequency, with the most frequent reactions first. Within each frequency grouping, adverse drug reactions are presented in order of decreasing seriousness. In addition, the corresponding frequency category for each adverse drug reaction is based on the following convention (CIOMS III): very common (≥1/10); common (≥1/100 to <1/10); uncommon (≥1/1,000 to <1/100); rare (≥1/10,000 to <1/1,000); very rare (<1/10,000). (See Table 15.)
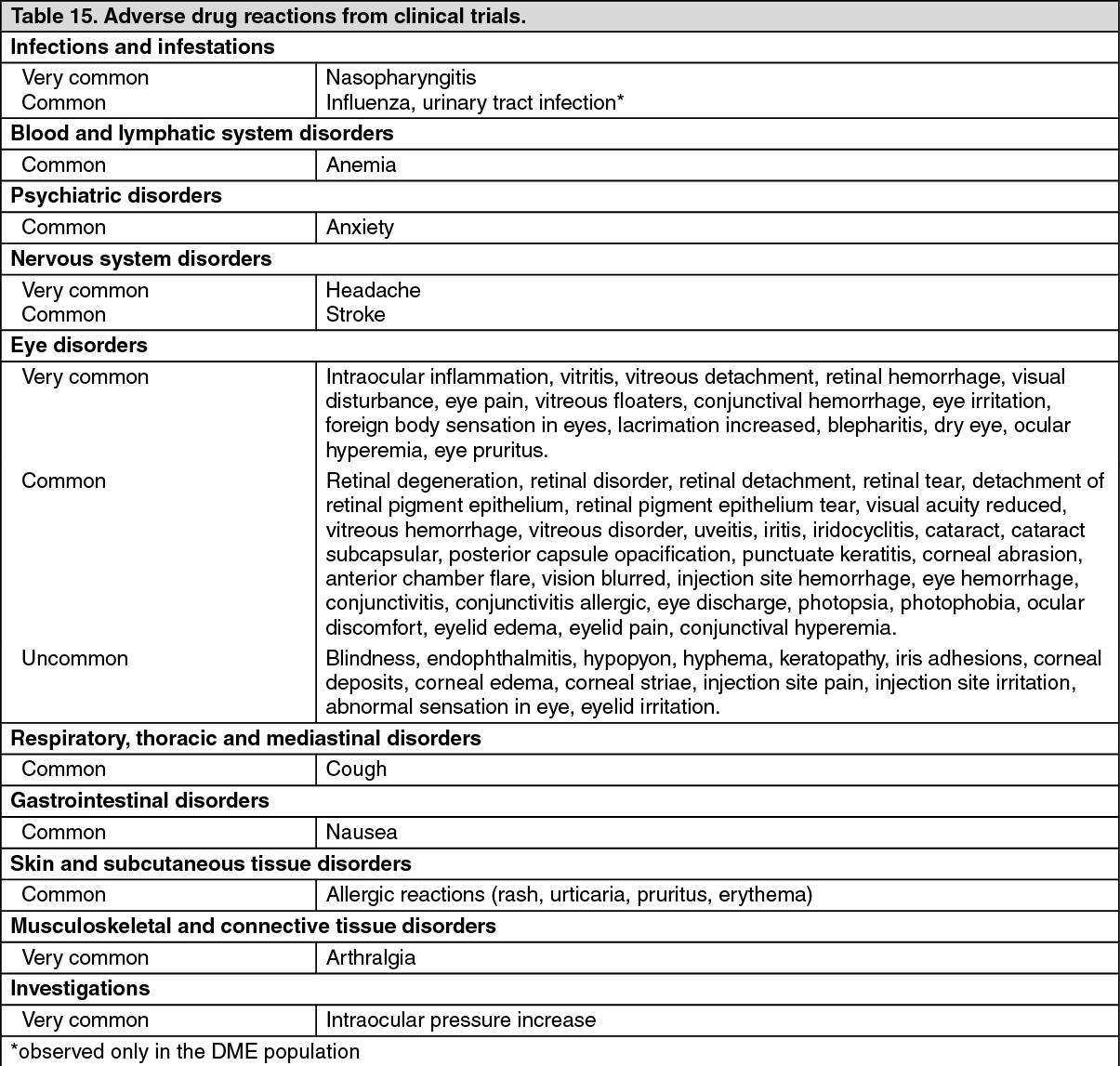 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageA meta-analysis of pooled safety data from completed, randomized, double masked global studies showed a higher incidence rate of non-serious, non-ocular wound infection/inflammation in DME patients treated with ranibizumab 0.5 mg (1.85/100 patient years) compared to control (0.27/100 patient years). The relationship to ranibizumab remains unknown.
Retinopathy of Prematurity (ROP) population: The safety of Accentrix 0.2 mg was studied in the 6-month clinical trial (RAINBOW), which included 73 ranibizumab-treated preterm infants with ROP (see Pharmacology: Pharmacodynamics: CLINICAL STUDIES under Actions). Ocular events observed in the RAINBOW trial were consistent with those seen in adults treated with ranibizumab 0.5 mg. In general, the non-ocular events in this trial were consistent with what would be expected for this patient population with multiple comorbidities due to prematurity.
View ADR Monitoring Form