Pharmacology: Pharmacodynamics: Mechanism of Action: Rituximab is a chimeric mouse/human monoclonal antibody that binds specifically to the transmembrane antigen, CD20. This antigen is located on pre-B- and mature B-lymphocytes, but not on haematopoietic stem cells, pro-B-cells, normal plasma cells or other normal tissue. The antigen is expressed on >95% of all B-cell non-Hodgkin's Lymphomas (NHLs). Following antibody binding, CD20 does not internalized or shed from the cell membrane into the environmen. CD20 does not circulate in the plasma as a free antigen and, thus, does not compete for antibody binding.
Rituximab binds to the CD20 antigen on B-lymphocytes and initiates immunologic reactions that mediate B-cell lysis. Possible mechanisms of cell lysis include complement-dependent cytotoxicity (CDC), antibody-dependent cellular cytotoxicity (ADCC), induction of apoptosis. Finally, in vitro studies have demonstrated that Rituximab sensitizes drug-resistant human B-cell lymphoma lines to the cytotoxic effects of some chemotherapeutic agents.
Peripheral B-cell counts declined to levels below normal following the first dose of Rituximab. In patients treated for hematological malignancies, B-cell recovery began within 6 months of treatment generally returning to normal levels within 12 months after completion of therapy, although in some patients this may take longer.
Rituximab Monotherapy: Initial treatment, weekly for 4 doses: In the pivotal study, 166 patients with relapsed or chemoresistant low-grade or follicular B-cell NHL received 375 mg/m
2 of Rituximab as an IV infusion weekly for four doses. The overall response rate (ORR) in the intent-to-treat (ITT) population was 48% (Cl95% 41%-56%) with a 6% complete response (CR) and a 42% partial response (PR) rate. The projected median time to progression (TTP) for responding patients was 13.0 months.
In a subgroup analysis, the ORR was higher in patients with IWF B, C, and D histologic subtypes as compared to IWF A subtype (58% vs. 12%), higher in patients whose largest lesion was < 5 cm versus > 7 cm in greatest diameter (53% vs. 38%), and higher in patients with chemosensitive relapse as compared to chemoresistant (defined as duration or response < 3 months) relapse (50% vs. 22%). ORR in patients previously treated with autologous bone marrow transplant (ABMT) was 78% vs. 43% in patients with no ABMT. Neither age, sex, lymphoma grade, initial diagnosis, presence or absence of bulky disease, normal or high LDH nor presence of extranodal disease had a statistically significant effect (Fisher's exact test) on response to Rituximab.
A statistically significant correlation was noted between response rates and bone marrow involvement. Forty percent of patients with bone marrow involvement responded compared to 59% of patients with no bone marrow involvement (p=0.0186). This finding was not supported by a step-wise logistic regression analysis in which the following factors were identified as prognostic factors: histological type, bcl-2 positivity at baseline, resistance to last chemotherapy and bulky disease.
Initial treatment, weekly for 8 doses: In a multi-centre, single-arm study, 37 patients with relapsed or chemoresistant, low grade or follicular B-cell NHL received 375 mg/m
2 of Rituximab as IV infusion weekly for eight doses. The ORR was 57% (Cl95% 41%-73%; CR 14%, PR 43%) with a projected median TTP for responding patients of 19.4 months (range 5.3 to 38.9 months).
Initial treatment, bulky disease, weekly for 4 doses: In pooled data from three studies, 39 patients with relapsed or chemoresistant, bulky disease (single lesion ≥10 cm in diameter), low grade or follicular B-cell NHL received 375 mg/m
2 of Rituximab as IV infusion weekly for four doses. The ORR was 36% (Cl95% 21%-51%; CR 3%, PR 33%) with a median TTP for responding patients of 9.6 months (range 4.5 to 26.8 months).
Re-treatment, weekly for 4 doses: In a multi-center, single-arm study, 58 patients with relapsed or chemorasistant low grade or follicular B-cell NHL, who had achieved an objective clinical response to a prior course of Rituximab were retreated with 375 mg/m
2 of Rituximab as IV infusion weekly for four doses.
Three of the patients had received two courses of Rituximab before enrollment and thus were given a third course in the study. Two patients were re-treated twice in the study. For the 60 retreatments on study, the ORR was 38% (Cl95% 26%-51%; 10% CR, 28% PR) with a projected median TTP for responding patients of 17.8 months (range 5.4-26.6). This compares favorably with the TTP achieved after the prior course of Rituximab (12.4 months).
Rituximb in Combination with Chemotherapy: Initial treatment: In an open-label randomized trial, a total of 322 previously untreated patients with follicular lymphoma were randomized to receive either CVP chemotherapy (Cyclophosphamide 750 mg/m
2, Vincristine 1.4 mg/m
2 up to a maximum of 2 mg on day 1, and Prednisolone 40 mg/m
2/day on days 1-5) every 3 weeks for 8 cycles or Rituximab 375 mg/m
2 in combination with CVP (R-CVP). Rituximab was administered on the first day of each treatment cycle. A total of 321 patients (162 R-CVP, 159 CVP) received therapy and were analyzed for efficacy.
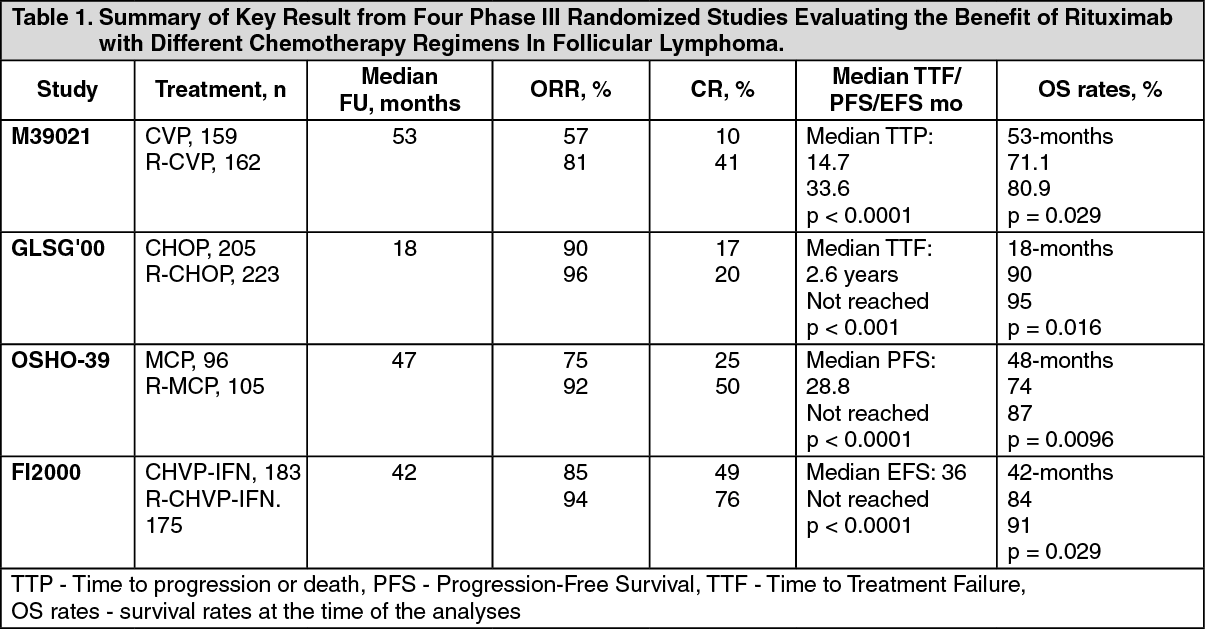
The median follow-up of patients was 53 months. R-CVP led lo a significant benefit over CVP for the primary end-point, time to treatment failure (27 months vs 6.6 months, p< 0.0001, log-rank test). The proportion of patients with a tumor response (CR, CRu, PR) was significantly higher (p <0.0001 Chi-Square test) in the R-CVP group (80.9%) than the CVP group (57.2%). Treatment with R-CVP significantly prolonged the time to disease progression or death compared to CVP, 33.6 months and 14.7 months, respectively (p < 0.0001, log-rank test). The median duration of response was 37.7 months in the R-CVP group and was 13.5 months in the CVP group (p < 0.0001, log-rank test). The difference between the treatment groups with respect to overall survival showed a strong clinical benefit (p=0.029, log-rank test stratified by center): survival rates at 53 months were 80.9% for patients in the R-CVP group compared to 71.1% for patients in the CVP group.
Results from three other randomized trials using Rituximab in combination with chemotherapy regimen other than CVP (CHOP, MCP, CHVP/lnterferon-α) also demonstrated significant improvements in response rates, time-dependent parameters as well as in overall survival. Key results from all four studies are summarized in Table 1 as follows. See Table 1.
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Rituximab Maintenance Therapy: Previously untreated follicular NHL: In a prospective, open label, international, multi-centre, phase III trial 1193 patients with previously untreated advanced follicular lymphoma received induction therapy with R-CHOP (n=881), RCVP (n=268) or R-FCM (n=44), according to the investigators choice. A total of 1078 patients responded to induction therapy, of which 1018 were randomized to Rituximab IV maintenance therapy (n=505) or observation (n=513). The two treatment groups were well balanced with regards to baseline characteristics and disease status. Rituximab maintenance treatment consisted of a single infusion of Rituximab at 375 mg/m
2 BSA given every 2 months until disease progression or for a maximum period of two years.
After a median observation time of 25 months for randomization, maintenance therapy with Rituximab resulted in a clinically relevant and statistically significant improvement in the primary endpoint of investigator assessed progression-free survival (PFS) as compared to no maintenance therapy in patients with previously untreated follicular NHL (Table 2). This improvement in PFS was confirmed by an independent review committee IRC) (see Table 2 as follows).
Significant benefit from maintenance treatment with Rituximab was also seen for the secondary endpoints event-free survival (EFS), time to next anti-lymphoma treatment (TNLT) time to next chemotherapy (TNCT) and overall response rate (ORR) (see Table 2 as follows). See Table 2.
Click on icon to see table/diagram/image
Rituximab maintenance treatment provided consistent benefit in all subgroups tested: gender (male, female), age (< 60 years, ≥60 years), FLIPI score (1, 2 or 3), induction therapy (R-CHOP, R-CVP or R-FCM) and regardless of the quality of response to induction treatment (CR or PR).
Relapsed/Refractory follicular NHL: In a prospective, open label, international, multicenter, phase III trial, 465 patients with relapsed/refractory follicular NHL were randomized in a first step to induction therapy with either CHOP (Cyclophosphamide, Doxorubicin, Vincristine, Prednisolone; n=231) or Rituximab plus CHOP (R-CHOP, n=234). The two treatment groups were well balanced with regard to baseline characteristics and disease status. A total of 334 patients achieving a complete or partial remission.
Following induction therapy were randomized in a second step to Rituximab maintenance therapy (n=167) or observation (n=167). Rituximab maintenance treatment consisted of a single infusion of Rituximab at 375 mg/m
2 BSA given every 3 months until disease progression or for a maximum period of two years.
The final efficacy analysis included all patients randomized to both parts of the study. After a median observation time of 31 months for patients randomized to the induction phase, R-CHOP significantly improved the outcome of patients with relapsed/refractory follicular NHL when compared to CHOP (see Table 3 as follows). See Table 3.
Click on icon to see table/diagram/image
For patients randomized to the maintenance phase of the trial, the median observation time was 28 months from maintenance randomization. Maintenance treatment with Rituximab led to a clinically relevant and statistically significant improvement in the primary end-point, PFS, (time from maintenance randomization to relapse, disease progression or death) when compared to observation alone (p< 0.0001 log-rank test). The median PFS was 42.2 months in the Rituximab maintenance arm compared to 14.3 months in the observation arm. Using a cox regression analysis, the risk of experiencing progressive disease or death was reduced by 61% with Rituximab maintenance treatment when compared to observation (95% Cl; 45%-72%). Kaplan-Meier estimated progression-free rates at 12 months were 78% in the Rituximab maintenance group vs. 57% in the observation group. An analysis of overall survival confirmed the significant benefit of Rituximab maintenance over observation (p = 0.0039 log-rank test). Rituximab maintenance treatment reduced the risk of death by 56% (95% Cl; 22%-75%).
The median time to new anti-lymphoma treatment was significantly longer with Rituximab maintenance treatment than with observation (38.8 months vs. 20.1 months, p <0.0001 log-rank test). The risk of starting a new treatment was reduced by 50% (95% Cl; 30%-64%). In patients achieving a CR/CRu (complete response unconfirmed) as best response during induction treatment, Rituximab maintenance treatment significantly prolonged the median disease free survival (DFS) compared to the observation group (53. 7 vs. 16.5 months, p = 0.0003 log-rank test) (see Table 4 as follows). The risk of relapse in complete responders was reduced by 67% (95% Cl: 39%-82%). See Table 4.
Click on icon to see table/diagram/image
The benefit of Rituximab maintenance treatment was confirmed in all subgroups analyzed, regardless of induction regimen (CHOP or R-CHOP) or quality of response to induction treatment (CR or PR) (see Table 4). Rituximab maintenance treatment significantly prolonged median PFS in patients responding to CHOP induction therapy (median PFS 37.5 months vs. 11.6 months, p < 0.0001) as well as in those responding to R-CHOP induction (median PFS 51.9 months vs. 22.1 months, p = 0.0071). Rituximab maintenance treatment also provided a clinically meaningful benefit in terms of overall survival for both patients responding to CHOP and patients responding to R-CHOP in the induction phase of the study.
Rituximab maintenance treatment provided consistent benefit in all subgroups tested: gender, age (60 years, >60 years), stage (III, IV), WHO performance status (0 vs. >0), B symptoms (absent, present), bone marrow involvement (no vs. yes), IPI (0-2 vs. 3-5), FLIPI score (0-1, vs. 3-5), number of extranodal sites (0-1 vs. >1), number of nodal sites (<5 vs. 5), number of previous regimens (1 vs. 2), best response to prior therapy (CR/PR vs. NC/PD), haemoglobin (<12 g/dL vs. 12 g/dL, β
2-microglobulin (< 3 mg/L vs. 3 mg/L), LDH (elevated, not elevated) except for the small subgroup of patients with bulky disease.
Diffuse Large B-cell Non-Hodgkin's Lymphoma: In a randomized, open-label trial, a total of 399 previously untreated elderly patients (age 60 to 80 years) with diffuse large B-cell lymphoma received standard CHOP chemotherapy (Cyclophosphamide 750 mg/m
2, Doxorubicin 50 mg/m
2, Vincristine 1.4 mg/m
2 up to a maximum of 2 mg on day 1, and Prednisolone 40 mg/m
2/day on days 1-5) every 3 weeks for eight cycles, or Rituximab 375 mg/m
2 plus CHOP (R-CHOP), Rituximab was administered on the first day of the treatment cycle.
The final efficacy analysis included all randomized patients (197 CHOP, 202 R-CHOP), and had a median follow-up duration of approximately 31 months. The two treatment groups were well balanced in baseline characteristics and disease status. The final analysis confirmed that R-CHOP significantly increased the duration of event-free survival (the primary efficacy parameter, where events were death, relapse or progression of lymphoma, or institution of a new anti-lymphoma treatment) (p = 0.0001). Kaplan Meier estimates of the median duration of event-free survival were 35 months in the R-CHOP arm compared to 13 months in the CHOP arm, representing a risk reduction of 41%. At 24 months, estimates for overall survival were 68.2% in the R-CHOP arm compared to 57.4% in the CHOP arm. A subsequent analysis of the duration of overall survival, carried out with a median follow-up duration of 60 months, confirmed the benefit of R-CHOP over CHOP treatment (p =0.0071), representing a risk reduction of 32%.
The analysis of all secondary parameters (response rates, progression-free survival, disease-free survival, duration of response) verified the treatment effect of R-CHOP compared to CHOP. The complete response rate after Cycle 8 was 76.2% in the R-CHOP group and 62.4% in the CHOP group (p = 0.0028). The risk of disease progression was reduced by 46% and the risk of relapse by 51%.
In all patients subgroups (gender, age, age adjusted IPI, Ann Arbor stage, ECOG, Beta 2 Microglobulin, LDH, Albumin, B-symptoms, Bulky disease, Extranodal sites, bone marrow involvement), the risk ratios for event-free survival and overall survival (R-CHOP compared with CHOP) were less than 0.83 and 0.95; respectively. R-CHOP was associated with improvements in outcome for both high- and low-risk patients according to age adjusted IPI.
Pharmacokinetics: Absorption: Not applicable.
Distribution: Non-Hodgkin's Lymphoma: Based on a population pharmacokinetic analysis in 298 NHL patients who received single or multiple infusions of Rituximab as a single agent or in combination with CHOP therapy, the typical population estimates of nonspecific clearance (CL1), specific clearance (CL2) likely contributed by B-cells or tumor burden, and central compartment volume of distribution (V1) were 0.14 L/day, 0.59 L/day, and 2.7 L, respectively. The estimated median terminal elimination half-life of Rituximab was 22 days (range, 6.1 to 52 days). Baseline CD19-positive cell counts and size of measurable tumor lesions contributed to some of the variability in CL2 of Rituximab in data from 161 patients given 375 mg/m
2 as an IV infusion for 4 weekly doses. Patients with higher CD19-positive cell counts or tumor lesions had a higher CL2. However, a large component of inter-individual variability remained for CL2 after correction for CD19-positive cell counts and tumor lesion size. V1 varied by body surface area (BSA) and CHOP therapy. This variability in V1 (27.1% and 19.0%) contributed by the range in BSA (1.53 to 2.32 m
2) and concurrent CHOP therapy, respectively, were relatively small. Age, gender, race, and WHO performance status had no effect on the pharmacokinetics of Rituximab. This analysis suggests that dose adjustment of Rituximab with any of the tested co-variates is not expected to result in a meaningful reduction in its pharmacokinetic variability.
Rituximab at a dose of 375 mg/m
2 was administered as an IV infusion at weekly intervals for 4 doses to 203 patients with NHL naive to Rituximab. The mean Cmax following the fourth infusion was 486 μg/mL (range, 77.5 to 996.6 μg/mL). The peak and trough serum levels of Rituximab were inversely correlated with baseline values for the number of circulating CD19-positive B-cells and measures of disease burden. Median steady-state serum levels were higher for responders compared with non-responders, Serum levels were higher in patients with International Working Formulation (IWF) sub-types B, C, and D as compared with those with sub-type A. Rituximab was detectable in the serum of patients 3-6 months after completion of last treatment.
Rituximab at a dose of 375 mg/m
2 was administered as an IV infusion at weekly intervals for 8 doses to 37 patients with NHL. The mean Cmax increased with each successive infusion, spanning from a mean of 243 μg/mL (range, 16-582 μg/mL) after the first infusion to 550 μg/mL (range, 171-1177 μg/mL) after the eighth infusion.
The pharmacokinetic profile of Rituximab when administered as 6 infusions of 375 mg/m
2 in combination with 6 cycles of CHOP chemotherapy was similar to that seen with Rituximab alone.
Metabolism: No information available.
Elimination: See Distribution.
Pharmacokinetics In Special Populations: No pharmacokinetic data are available in patients with hepatic or renal impairment.
Usage In Special Populations: Pregnancy: There are no adequate and well-controlled studies of Rituximab in pregnant women. Women of child-bearing potential should use effective contraception while receiving Rituximab and for 12 months following treatment. Rituximab should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Human Data: Post-marketing data indicated that B-cell lymphocytopenia generally lasting less than six months can occur in infants exposed to Rituximab in-utero. Rituximab was detected postnatally in the serum of infants exposed in-utero.
Animal Data: In an embryo-fetal developmental toxicity study performed on pregnant cynomolgus monkeys, Rituximab crosses the monkey placenta. Exposed offspring did not exhibit any teratogenic effects but did have decreased B-cells and immunosuppression were noted in the offspring of Rituximab treated pregnant animals in subsequent pre- and post-natal reproductive toxicity studies and the B-cell counts returned to a normal levels, and immunologic function was restored within 6 months postpartum.
Nursing Mothers: It is not known whether Rituximab is secreted into human milk. However, Rituximab is secreted in the milk of lactating cynomolgus monkeys, and IgG is secreted in human milk. Published data suggest that antibodies in breast milk do not enter the neonatal and infant circulations in substantial amounts. The unknown risk to the infants from oral ingestion of Rituximab should be weighed against the known benefits of breastfeeding.
Pediatric Use: The safety and effectiveness of Rituximab in pediatric patients have not been established.
Geriatric Use: No information available.
Toxicology: Preclinical Safety: Carcinogenicity: No long-term animal studies have been performed to establish the carcinogenic potential of Rituximab, or to determine its effects on fertility in males or females. Standard tests to investigate mutagenicity have not been carried out, since such tests are not relevant for this molecule. However, due to its character it is unlikely that Rituximab has any mutagenic potential.
Mutagenicity: No information available.
Impairment of Fertility: No information available.
Teratogenicity: No information available.
Other: Rituximab has shown to be highly specific to the CD20 antigen on B-cells. Toxicity studies in cynomolgus monkeys have shown no other effect than the expected pharmacological depletion of B-cells in peripheral blood and in lymphoid tissue.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image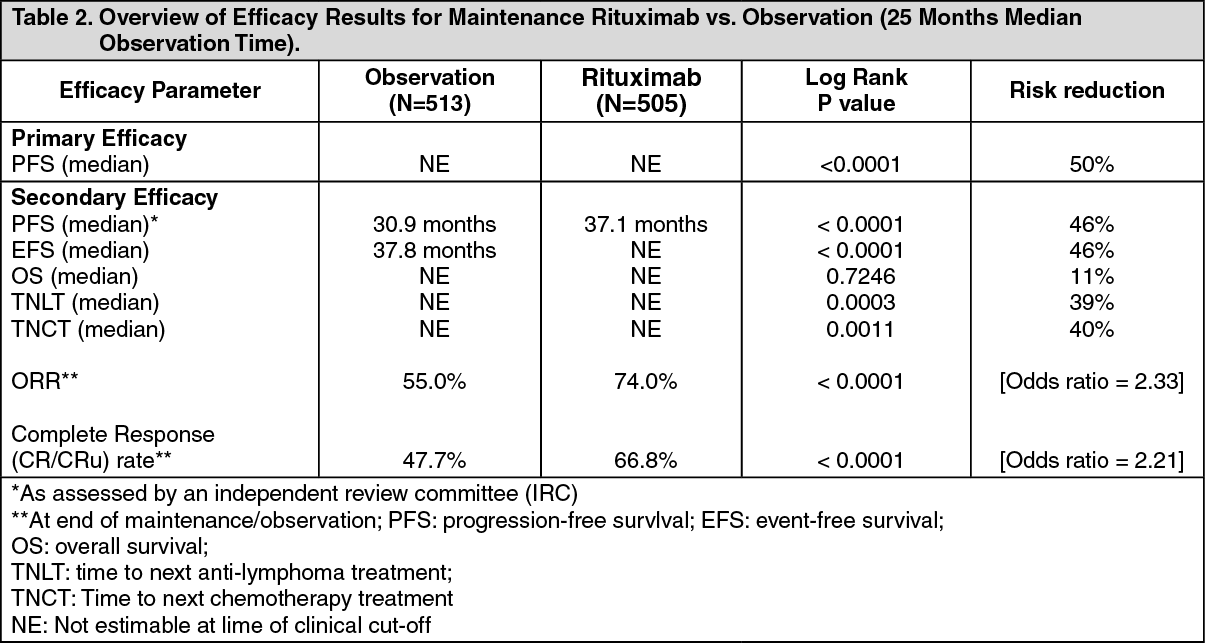 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image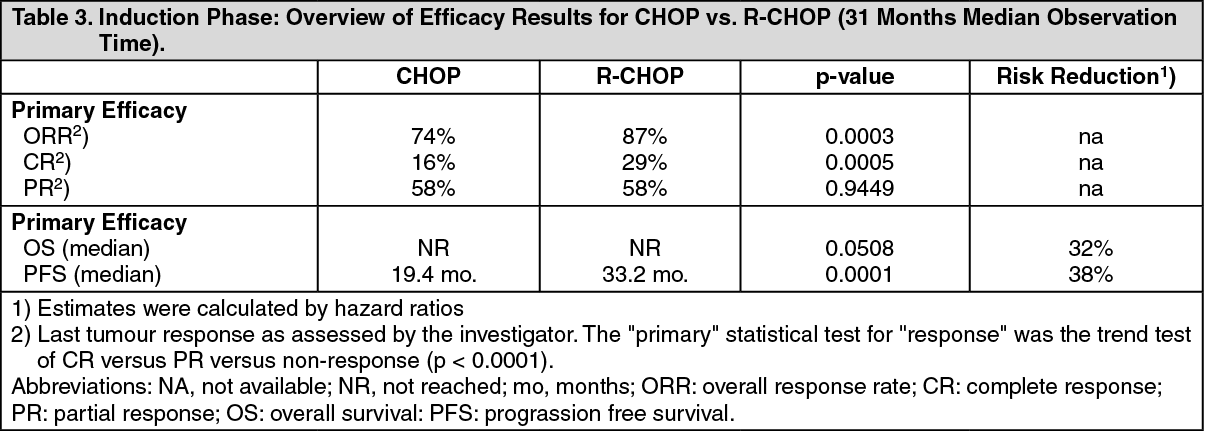 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image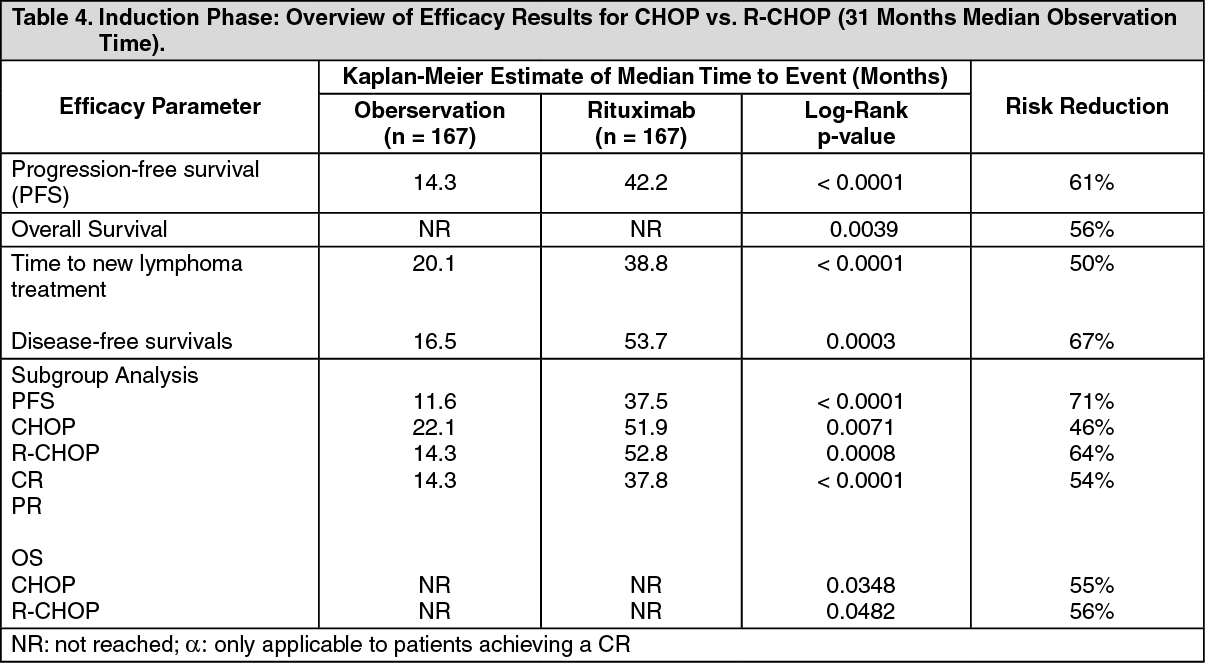 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
