


 Sign Out
Sign Out



Pharmacodynamic effects: Through inhibition of CDK4/6, palbociclib reduced cellular proliferation by blocking progression of the cell from G1 into S phase of the cell cycle. Testing of palbociclib in a panel of molecularly profiled breast cancer cell lines revealed high efficacy against luminal breast cancers, particularly estrogen receptor (ER)-positive breast cancers. In the cell lines tested, the loss of retinoblastoma (Rb) was associated with loss of palbociclib activity. Available clinical data are reported in the clinical efficacy and safety section (see Pharmacodynamics as previously mentioned). Mechanistic analyses revealed that the combination of palbociclib with anti-estrogen agents enhanced the re-activation of retinoblastoma (Rb) through inhibition of Rb phosphorylation resulting in reduced E2F signaling and growth arrest. In vivo studies using a patient derived ER-positive breast cancer xenograft model (HBCx-34) demonstrated that the combination of palbociclib and letrozole further enhanced inhibition of Rb phosphorylation, downstream signaling, and dose-dependent tumor growth. Studies are ongoing investigating the importance of Rb expression for the activity of palbociclib in fresh tumour samples.
Cardiac electrophysiology: The effect of palbociclib on the QT interval corrected for heart rate (QTc) interval was evaluated using time matched electrocardiogram (ECG) evaluating the change from baseline and corresponding pharmacokinetic data in 77 patients with advanced breast cancer. Palbociclib did not prolong the QTc to any clinically relevant extent at the recommended dose of 125 mg daily (Schedule 3/1).
Clinical trial efficacy: Study 1: The efficacy of palbociclib was evaluated in a randomized, open-label, multicenter study of palbociclib plus letrozole versus letrozole alone conducted in postmenopausal women with ER-positive, HER2-negative locally advanced or metastatic breast cancer.
The study was comprised of a limited Phase 1 portion (N = 12), designed to confirm the safety and tolerability of the combination palbociclib plus letrozole, followed by a randomized Phase 2 portion (N = 165), designed to evaluate the efficacy and safety of palbociclib in combination with letrozole compared with letrozole alone in the first-line treatment of postmenopausal women with ER-positive, HER2-negative advanced breast cancer.
Randomization was stratified by disease site (visceral versus bone only versus other) and by disease-free interval (>12 months from the end of adjuvant treatment to disease recurrence versus ≤12 months from the end of adjuvant treatment to disease recurrence or de novo advanced disease).
The patient demographic and baseline characteristics were generally balanced between the study arms in terms of age, race, disease sites, stage, and prior therapies.
The primary endpoint of the study was investigator-assessed progression-free survival (PFS) evaluated according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.0.
The median PFS (mPFS) for patients in the palbociclib plus letrozole arm was 20.2 months (95% confidence interval [CI]: 13.8, 27.5) and 10.2 months (95% CI: 5.7, 12.6) for patients in the letrozole-alone arm. The observed hazard ratio (HR) was 0.488 (95% CI: 0.319, 0.748) in favor of palbociclib plus letrozole, with a stratified log-rank test 1-sided p-value of 0.0004.
At the final overall survival (OS) analysis, the observed HR was 0.897 (95% CI: 0.623, 1.294) with a stratified 1-sided p-value of 0.2812. The median OS in the palbociclib plus letrozole arm was 37.5 months (95% CI: 31.4, 47.8) and in the letrozole alone arm was 34.5 months (95% CI: 27.4, 42.6).
The estimated survival probabilities at 12, 24, and 36 months between the 2 treatment arms were 89.0% versus 87.0%, 77.9% versus 71.1%, and 50.8% versus 47.4%, in favor of palbociclib plus letrozole, respectively.
Study 2: The efficacy of palbociclib in combination with letrozole versus letrozole plus placebo was evaluated in an international, randomized, double-blind, placebo-controlled, parallel-group, multicenter study conducted in women with ER-positive, HER2-negative locally advanced or metastatic breast cancer (PALOMA-2) who had not received prior systemic treatment for their advanced disease.
A total of 666 postmenopausal women were randomized 2:1 to either the palbociclib plus letrozole arm or to the placebo plus letrozole arm and were stratified by site of disease (visceral, non-visceral), disease-free interval from the end of (neo)adjuvant treatment to disease recurrence (de novo metastatic, ≤12 months from the end of adjuvant treatment to disease recurrence, >12 months from the end of adjuvant treatment to disease recurrence), and by the type of prior (neo)adjuvant anticancer therapies (prior hormonal therapy, no prior hormonal therapy). Patients with advanced symptomatic, visceral spread, that were at risk of life-threatening complications in the short term (including patients with massive uncontrolled effusions [pleural, pericardial, peritoneal], pulmonary lymphangitis, and over 50% liver involvement), were not eligible for enrolment into the study.
Patients continued to receive their assigned treatment until objective disease progression, symptomatic deterioration, unacceptable toxicity, death, or withdrawal of consent, whichever occurred first. Crossover between treatment arms was not allowed.
Patients were well matched for baseline demographics and disease characteristics between the palbociclib plus letrozole arm and the placebo plus letrozole arm. The median age of patients enrolled in this study was 62 years (range 28-89); 48.3% of patients had received chemotherapy and 56.3% had received antihormonal therapy in the (neo)adjuvant setting prior to their diagnosis of advanced breast cancer, while 37.2% of patients had received no prior systemic therapy in the (neo)adjuvant setting. Most patients (97.4%) had metastatic disease at baseline; 22.7% of patients had bone only disease and 49.2% of patients had visceral disease.
The primary endpoint of the study was PFS evaluated according to RECIST version 1.1 as assessed by investigator. Secondary efficacy endpoints included objective response (OR), duration of response (DOR), clinical benefit response (CBR), overall survival (OS), safety, EQ-5D scores and health-related quality of life (QoL) assessed using the FACT-B questionnaire.
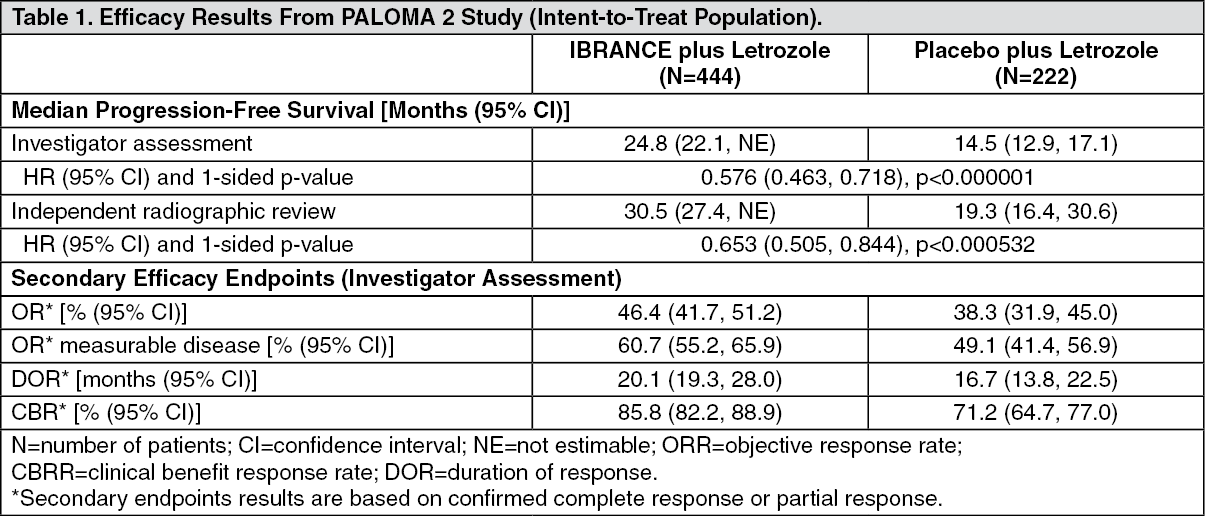
The study met its primary objective of improving PFS. The estimated HR was 0.576 (95% CI: 0.463, 0.718) in favor of palbociclib plus letrozole, with a stratified log-rank test 1-sided p-value of <0.000001. The mPFS for patients in the palbociclib plus letrozole arm was 24.8 months (95% CI: 22.1, Not estimable [NE]) and 14.5 months (95% CI: 12.9, 17.1) for patients in the placebo plus letrozole arm. The treatment effect of the combination on PFS was also supported by an independent review of radiographs with an estimated HR of 0.653 (95% CI: 0.505, 0.844).
Efficacy data from the PALOMA-2 study are summarized in Table 1 and the Kaplan-Meier curve for PFS is shown in the figure. (See Table 1 and figure.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image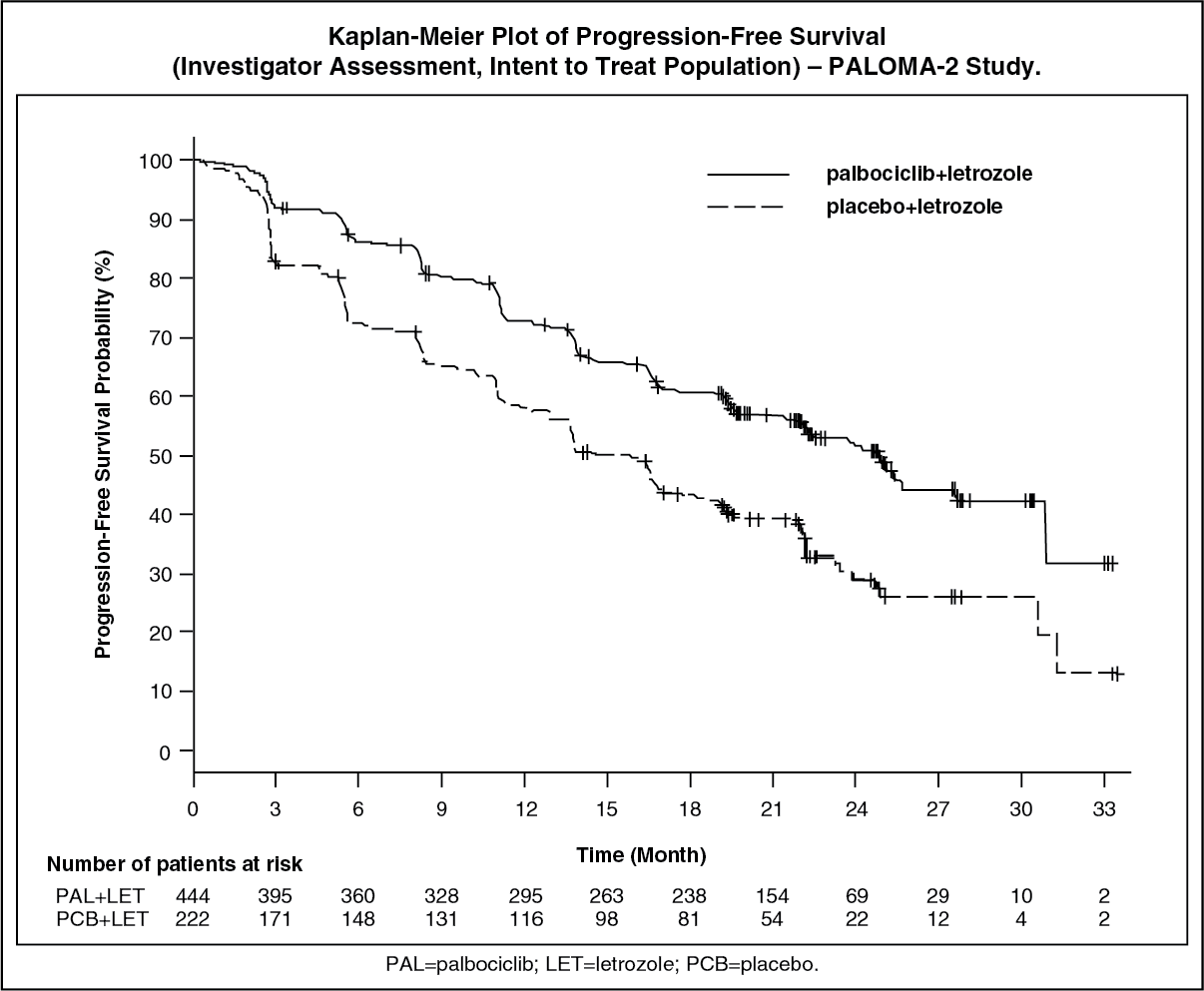 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageA series of prespecified subgroup PFS analyses was performed based on baseline demographic and disease characteristics to investigate the internal consistency of treatment effect. A reduction in the risk of disease progression or death in the palbociclib plus letrozole arm was observed in all individual patient subgroups defined by stratification factors and baseline characteristics in the primary and in the updated analyses. Patients with advanced, symptomatic visceral spread at risk of life-threatening complications in the short term were not included in PALOMA-2. Objective Response Rate (ORR) was higher in patients with visceral disease treated with palbociclib plus letrozole compared to placebo plus letrozole (58.9% [95% CI: 52.0, 65.5], versus 45.5% [95% CI: 35.9, 55.2], respectively), while in patients with non visceral metastases (n= 342, of whom 151 had bone only disease) ORR was comparable between treatment groups (34.8% [95% CI: 28.6, 41.3], versus 31.3% [95% CI: 22.8, 40.7]) The overall survival (OS) data were not mature at the time of the final PFS analysis (20% of patients had died). Patients will continue to be followed for the final analysis.
An analysis of time-to-deterioration composite endpoint (TTD) in Functional Assessment of Cancer Therapy-Breast (FACT-B), defined as the time between baseline and first occurrence of decrease of ≥7 points in FACT-B scores, was carried out based on survival analysis methods using a Cox proportional hazards model and log-rank test. No statistically significant difference was observed in TTD in FACT-B total scores between the palbociclib plus letrozole arm and the placebo plus letrozole arm (HR of 1.042 [95% CI: 0.838, 1.295]; 1-sided p-value=0.663.
Pharmacokinetics: The pharmacokinetics of palbociclib were characterized in patients with solid tumors including advanced breast cancer and in healthy subjects.
Absorption: The mean time to Cmax (Tmax) of palbociclib is generally between 6 to 12 hours following oral administration. The mean absolute bioavailability of palbociclib after an oral 125 mg dose is 46%. In the dosing range of 25 mg to 225 mg, the AUC and Cmax increase proportionally with dose in general. Steady state was achieved within 8 days following repeated once daily dosing. With repeated once daily administration, palbociclib accumulates with a median accumulation ratio of 2.4 (range 1.5-4.2).
Food effect: Palbociclib absorption and exposure were very low in approximately 13% of the population under the fasted condition. Food intake increased the palbociclib exposure in this small subset of the population, but did not alter palbociclib exposure in the rest of the population to a clinically relevant extent. Therefore, food intake reduced the intersubject variability of palbociclib exposure, which supports administration of IBRANCE with food.
Compared to palbociclib given under overnight fasted conditions, the AUCinf and Cmax of palbociclib increased by 21% and 38% when given with high-fat food, by 12% and 27% when given with low-fat food, and by 13% and 24% when moderate-fat food was given 1 hour before and 2 hours after palbociclib dosing. In addition, food intake significantly reduced the intersubject and intrasubject variability of palbociclib exposure. Based on these results, palbociclib should be taken with food.
Gastric pH elevating medication effect: In a healthy subject study, coadministration of a single 125 mg dose of IBRANCE with multiple doses of the PPI rabeprazole under fed conditions decreased palbociclib Cmax by 41%, but had limited impact on AUCinf (13% decrease), when compared to a single 125 mg dose of IBRANCE administered alone. Given the reduced effect on gastric pH of H2 receptor antagonists and local antacids compared to PPIs, the effect of these classes of acid-reducing agents on palbociclib exposure under fed conditions is expected to be minimal. Under fed conditions there is no clinically relevant effect of PPIs, H2-receptor antagonists, or local antacids on palbociclib exposure. In another healthy subject study, coadministration of a single 125 mg dose of IBRANCE with multiple doses of the PPI rabeprazole under fasted conditions decreased palbociclib AUCinf and Cmax by 62% and 80%, respectively, when compared with a single dose of IBRANCE administered alone.
Distribution: Binding of palbociclib to human plasma proteins in vitro was ~85%, with no concentration dependence over the concentration range of 500 ng/mL to 5000 ng/mL. The mean fraction unbound (fu) of palbociclib in human plasma in vivo increased incrementally with worsening hepatic function. There was no obvious trend in the mean palbociclib fu in human plasma in vivo with worsening renal function. The geometric mean apparent volume of distribution (Vz/F) was 2583 (26%) L.
Metabolism: In vitro and in vivo studies indicate that palbociclib undergoes extensive hepatic metabolism in humans. Following oral administration of a single 125 mg dose of [14C] palbociclib to humans, the major primary metabolic pathways for palbociclib involved oxidation and sulfonation, with acylation and glucuronidation contributing as minor pathways. Palbociclib was the major circulating drug-derived entity in plasma. The major circulating metabolite was a glucuronide conjugate of palbociclib, although it only represented 1.5% of the administered dose in the excreta. The majority of the material was excreted as metabolites. In feces, the sulfamic acid conjugate of palbociclib was the major drug-related component, accounting for 25.8% of the administered dose. In vitro studies with human hepatocytes, liver cytosolic and S9 fractions, and recombinant sulfotransferase (SULT) enzymes indicated that CYP3A and SULT2A1 are mainly involved in the metabolism of palbociclib.
Elimination: The geometric mean apparent oral clearance (CL/F) of palbociclib was 63.08 L/h, and the mean plasma elimination half-life was 28.8 hours in patients with advanced breast cancer. In 6 healthy male subjects given a single oral dose of [14C] palbociclib, a median of 91.6% of the total administered radioactive dose was recovered in 15 days; feces (74.1% of dose) was the major route of excretion, with 17.5% of the dose recovered in urine. Excretion of unchanged palbociclib in feces and urine was 2.3% and 6.9% of the administered dose, respectively.
Age, gender, and body weight: Based on a population pharmacokinetic analysis in 183 patients with cancer (50 male and 133 female patients, age ranging from 22 to 89 years, and body weight ranging from 37.9 to 123 kg), gender had no effect on the exposure of palbociclib, and age and body weight had no clinically important effect on the exposure of palbociclib.
Pediatric population: Pharmacokinetics of palbociclib have not been evaluated in patients ≤18 years of age.
Elderly population: Of 444 patients who received IBRANCE in Study A5481008 (PALOMA-2), 181 patients (41%) were ≥65 years of age with 133 (30%) patients between the age of 65 and 74, and 48 (11%) patients ≥75 years of age. Of 347 patients who received IBRANCE in Study A5481023 (PALOMA-3), 86 patients (25%) were ≥65 years of age with 59 (17%) patients between the age of 65 and 74, and 27 (8%) patients ≥75 years of age. No overall differences in safety were observed across all age groups and elderly age groups. Neutropenia was the most common adverse event with palbociclib across all age groups; however, the incidence of febrile neutropenia was low in all age groups.
Hepatic impairment: Data from a pharmacokinetic trial in subjects with varying degrees of hepatic function indicate that palbociclib unbound exposure (unbound AUCinf) decreased by 17% in subjects with mild hepatic impairment (Child-Pugh class A), and increased by 34% and 77% in subjects with moderate (Child-Pugh class B) and severe hepatic impairment (Child-Pugh class C), respectively, relative to subjects with normal hepatic function. Peak palbociclib unbound exposure (unbound Cmax) was increased by 7%, 38% and 72% for mild, moderate and severe hepatic impairment, respectively, relative to subjects with normal hepatic function. In addition, based on a population pharmacokinetic analysis that included 183 patients with advanced cancer where 40 patients had mild hepatic impairment based on National Cancer institute (NCI) classification (total bilirubin ≤Upper Limit of Normal (ULN) and Aspartate Aminotransferase (AST) >ULN, or total bilirubin >1.0 to 1.5 x ULN and any AST), mild hepatic impairment had no effect on the pharmacokinetics (PK) of palbociclib.
Renal impairment: Data from a pharmacokinetic trial in subjects with varying degrees of renal function indicate that total palbociclib exposure (AUCinf) was increased by 39%, 42%, and 31% with mild (60 mL/min≤CrCl<90 mL/min), moderate (30 mL/min≤CrCl<60 mL/min), and severe (CrCl <30 mL/min) renal impairment, respectively, relative to subjects with normal (CrCl ≥90 mL/min) renal function. Peak palbociclib exposure (Cmax) was increased by 17%, 12%, and 15% for mild, moderate, and severe renal impairment, respectively, relative to subjects with normal renal function. In addition, based on a population pharmacokinetic analysis that included 183 patients with advanced cancer where 73 patients had mild renal impairment and 29 patients had moderate renal impairment, mild and moderate renal impairment had no effect on the PK of palbociclib. The pharmacokinetics of palbociclib have not been studied in patients requiring hemodialysis.
Asian race: In a dedicated PK study in healthy volunteers, palbociclib geometric mean AUCinf and Cmax values were 30% and 35% higher, respectively, in Japanese subjects compared with non-Asian subjects after a single oral dose. However, this finding was not reproduced consistently in subsequent studies in breast cancer patients after multiple dosing. Based on an analysis of the cumulative pharmacokinetic, safety, and efficacy data, no dose adjustment based on Asian race is necessary.
Cardiac electrophysiology: The effect of palbociclib on the QT interval corrected for heart rate (QTc) was evaluated using time-matched electrocardiograms (ECGs) evaluating the change from baseline and corresponding pharmacokinetic data in 77 patients with breast cancer. Palbociclib did not prolong QTc to any clinically relevant extent at the recommended dose of 125 mg daily (Schedule 3/1).
Toxicology: Preclinical safety data: The primary target organ findings following single and/or repeat dosing included hematolymphopoietic and male reproductive organ effects in rats and dogs, and effects on bone and actively growing incisors in rats only. These systemic toxicities were generally observed at clinically relevant exposures based on AUC. Partial to full reversal of effects on the hematolymphopoietic, male reproductive systems, and incisor teeth were established, whereas the bone effect was not reversed following a 12-week nondosing period. In addition, cardiovascular effects (QTc prolongation, decreased heart rate, and increased RR interval and systolic blood pressure) were identified in telemetered dogs at ≥4 times human clinical exposure based on Cmax.
Carcinogenicity: The relevance of the male rat neoplastic finding to humans is unknown.
Genotoxicity: Palbociclib was not mutagenic in a bacterial reverse mutation (Ames) assay and did not induce structural chromosomal aberrations in the in vitro human lymphocyte chromosome aberration assay.
Palbociclib induced micronuclei via an aneugenic mechanism in Chinese Hamster Ovary cells in vitro and in the bone marrow of male rats at doses ≥100 mg/kg/day. The no observed effect level for aneugenicity was approximately 7 times human clinical exposure based on AUC.
Impairment of fertility: Palbociclib did not affect mating or fertility in female rats at any dose tested up to 300 mg/kg/day (approximately 3 times human clinical exposure based on AUC) and no adverse effects were observed in female reproductive tissues in repeat-dose toxicity studies up to 300 mg/kg/day in the rat and 3 mg/kg/day in the dog (approximately 5 and 3 times human clinical exposure based on AUC, respectively).
Palbociclib is considered to have the potential to impair reproductive function and fertility in male humans based on nonclinical findings in rats and dogs. Palbociclib-related findings in the testis, epididymis, prostate, and seminal vesicle included decreased organ weight, atrophy or degeneration, hypospermia, intratubular cellular debris, lower sperm motility and density, and decreased secretion. These findings were observed in rats and/or dogs at exposures ≥9 times or subtherapeutic compared to human clinical exposure based on AUC. Partial reversibility of male reproductive organ effects was observed in the rat and dog following a 4- and 12-week non-dosing period, respectively. Despite these male reproductive organ findings, there were no effects on mating or fertility in male rats at projected exposure levels 13 times human clinical exposure based on AUC.
Developmental toxicity: Palbociclib was fetotoxic in pregnant animals. An increased incidence of a skeletal variation (increased incidence of a rib present at the seventh cervical vertebra) at ≥100 mg/kg/day was observed in rats. Reduced fetal body weights were observed at a maternally toxic dose of 300 mg/kg/day in rats (3 times human clinical exposure based on AUC), and an increased incidence of skeletal variations, including small phalanges in the forelimb was observed at a maternally toxic dose of 20 mg/kg/day in rabbits (4 times human clinical exposure based on AUC). Actual fetal exposure and cross-placenta transfer have not been examined.