Pharmacotherapeutic Group: Antineoplastic agents, monoclonal antibodies.
ATC Code: L01XC06.
Pharmacology: Pharmacodynamics: Mechanism of Action: Cetuximab is a chimeric monoclonal IgG
1 antibody that is specifically directed against the epidermal growth factor receptor (EGFR).
EGFR signalling pathways are involved in the control of cell survival, cell cycle progression, angiogenesis, cell migration and cellular invasion/metastasis.
Cetuximab binds to the EGFR with an affinity that is approximately 5- to 10-fold higher than that of endogenous ligands. Cetuximab blocks binding of endogenous EGFR ligands resulting in inhibition of the function of the receptor. It further induces the internalisation of EGFR, which can lead to down-regulation of EGFR. Cetuximab also targets cytotoxic immune effector cells towards EGFR-expressing tumour cells [antibody dependent cell-mediated cytotoxicity (ADCC)].
Cetuximab does not bind to other receptors belonging to the hydroxyethylrutosides (HER) family.
The protein product of the proto-oncogene Kirsten rat sarcoma 2 viral oncogene homologue (KRAS) is a central down-stream signal-transducer of EGFR. In tumours, activation of KRAS by EGFR contributes to EGFR-mediated increased proliferation, survival and the production of proangiogenic factors.
KRAS is one of the most frequently activated oncogenes in human cancers. Mutations of the KRAS gene at certain hot-spots (mainly codons 12 and 13) result in constitutive activation of the KRAS protein independently of EGFR signalling.
Pharmacodynamic Effects: In both
in vitro and
in vivo assays, cetuximab inhibits the proliferation and induces apoptosis of human tumour cells that express EGFR.
In vitro cetuximab inhibits the production of angiogenic factors by tumour cells and blocks endothelial cell migration.
In vivo cetuximab inhibits expression of angiogenic factors by tumour cells and causes a reduction in tumour neovascularisation and metastasis.
Immunogenicity: The development of human anti-chimeric antibodies (HACA) is a class effect of monoclonal chimeric antibodies. Current data on the development of HACAs is limited. Overall, measurable HACA titres were noted in 3.4% of the patients studied, with incidences ranging from 0-9.6% in the target indication studies. No conclusive data on the neutralising effect of HACAs on cetuximab is available to date. The appearance of HACA did not correlate with the occurrence of hypersensitivity reactions or any other undesirable effect to cetuximab.
Colorectal Cancer: A diagnostic assay (EGFR pharmDx) was used for immunohistochemical detection of EGFR expression in tumour material. A tumour was considered to be EGFR-expressing if 1 stained cell could be identified. Approximately 75% of the patients with metastatic colorectal cancer screened for clinical studies had an EGFR-expressing tumour and were therefore considered eligible for cetuximab treatment. The efficacy and safety of cetuximab have not been documented in patients with tumours where EGFR was not detected.
In metastatic colorectal cancer, the incidence of KRAS mutations is in the range of 30-50%. Recent data demonstrate that patients with KRAS wild-type metastatic colorectal cancer have a significantly higher chance to benefit from treatment with cetuximab or a combination of cetuximab and chemotherapy.
Cetuximab, as a single agent or in combination with chemotherapy, was investigated in 5 randomised controlled clinical studies and several supportive studies. The 5 randomised studies investigated a total of 3734 patients with metastatic colorectal cancer, in whom EGFR expression was detectable and who had an Eastern Cooperative Oncology Group (ECOG) performance status of ≤2. The majority of patients included had an ECOG performance status of ≤1. In all studies, cetuximab was administered as described in Dosage & Administration.
The KRAS status was recognised as predictive factor for the treatment with cetuximab in 4 of the randomised controlled studies. KRAS mutational status was available for 2072 patients. An analysis was not possible in study EMR 62 202-207 alone.
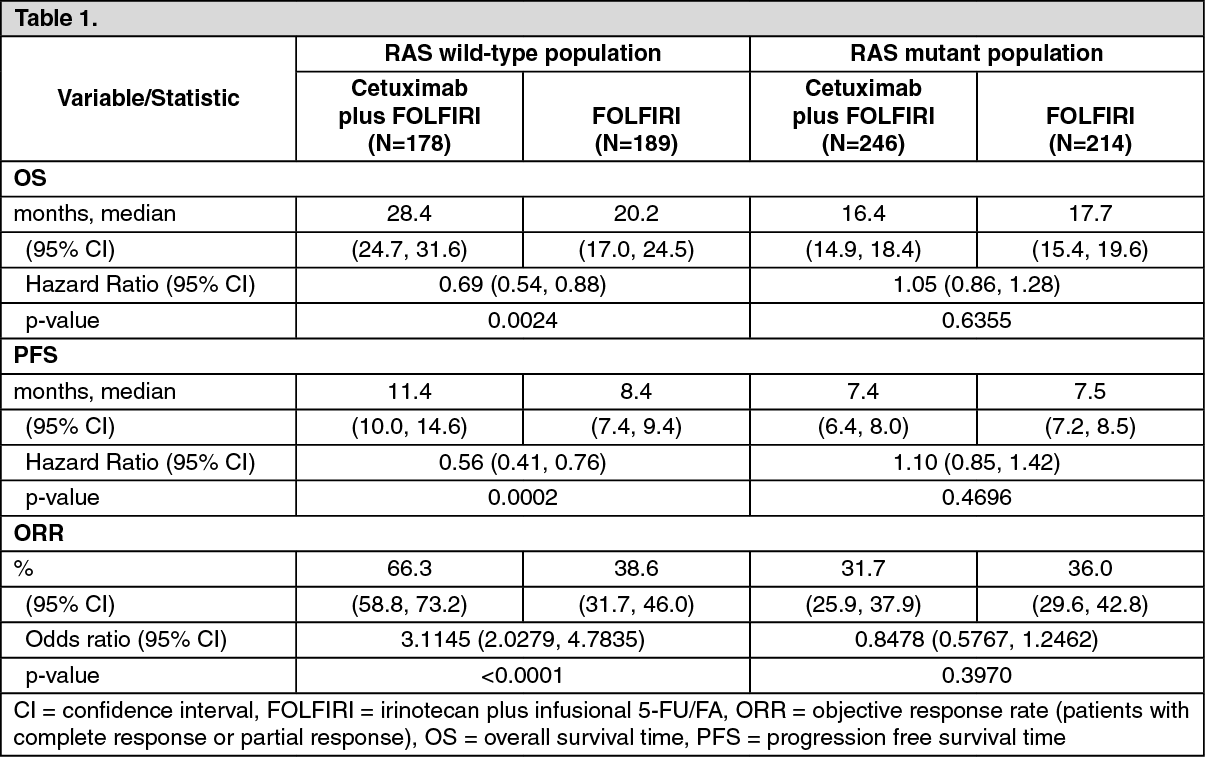
Cetuximab in Combination with Chemotherapy: EMR 62 202-013: This randomised study in patients with metastatic colorectal cancer who had not received prior treatment for metastatic disease compared the combination of cetuximab and irinotecan plus infusional 5-fluorouracil/folinic acid (FOLFIRI) (599 patients) to the same chemotherapy alone (599 patients). The proportion of patients with KRAS wild-type tumours from the patient population evaluable for KRAS status comprised 63%.
The efficacy data generated in this study are summarised in Table 1.
 Click on icon to see table/diagram/image
EMR 62 202-047:
Click on icon to see table/diagram/image
EMR 62 202-047: This randomised study in patients with metastatic colorectal cancer who had not received prior treatment for metastatic disease compared the combination of cetuximab and oxaliplatin plus infusional 5-fluorouracil/folinic acid (FOLFOX4) (169 patients) to the same chemotherapy alone (168 patients). The proportion of patients with KRAS wild-type tumours from the patient population evaluable for KRAS status comprised 57%.
The efficacy data generated in this study are summarised in Table 2.
Click on icon to see table/diagram/image
CA225006: This randomised study in patients with metastatic colorectal cancer who had received initial combination treatment with oxaliplatin plus fluoropyrimidine for metastatic disease compared the combination of cetuximab and irinotecan (648 patients) with irinotecan alone (650 patients). The proportion of patients with KRAS wild-type tumours from the patient population evaluable for KRAS status comprised 64%.
A significant difference in overall survival time could not be shown in this study. Following disease progression, treatment with EGFR-targeting agents was initiated in 50% of patients in the irinotecan-alone arm, which most likely impacted survival results. Objective response rate and progression free survival time were significantly improved with cetuximab. However, as no independent review of imaging data was conducted, these results have to be interpreted with caution.
EMR 62 202-007: This randomised study in patients with metastatic colorectal cancer after failure of irinotecan-based treatment for metastatic disease as the last treatment before study entry compared the combination of cetuximab and irinotecan (218 patients) with cetuximab monotherapy (111 patients).
The combination of cetuximab with irinotecan compared to cetuximab alone reduced the overall risk of disease progression by 46% and significantly increased objective response rate. In the randomised trial, the improvement in overall survival time did not reach statistical significance; however, in the follow-up treatment, nearly 50% of the patients of the cetuximab-alone arm received a combination of cetuximab and irinotecan after progression of disease, which may have influenced overall survival time.
Cetuximab as a Single Agent: CA225025: This randomised study in patients with metastatic colorectal cancer who had received prior oxaliplatin-, irinotecan- and fluoropyrimidine-based treatment for metastatic disease compared the addition of cetuximab as a single agent to best supportive care (BSC) (287 patients) with best supportive care (285 patients). The proportion of patients with KRAS wild-type tumours from the patient population evaluable for KRAS status comprised 58%.
The efficacy data generated in this study are summarised in Table 3.
Click on icon to see table/diagram/image
Squamous Cell Cancer of the Head and Neck: Immunohistochemical detection of EGFR expression was not performed since >90% of patients with squamous cell cancer of the head and neck have tumours that express EGFR.
Cetuximab in Combination with Radiation Therapy for Locally Advanced Disease: EMR 62 202-006: This randomised study compared the combination of cetuximab and radiation therapy (211 patients) with radiation therapy alone (213 patients) in patients with locally advanced squamous cell cancer of the head and neck. Cetuximab was started 1 week before radiation therapy and administered at the doses described in Dosage & Administration until the end of the radiation therapy period.
The efficacy data generated in this study are summarised in Table 4.
Click on icon to see table/diagram/image
Patients with a good prognosis as indicated by tumour stage, Karnofsky performance status (KPS) and age had a more pronounced benefit, when cetuximab was added to radiation therapy. No clinical benefit could be demonstrated in patients with KPS ≤80 who were ≥65 years.
The use of cetuximab in combination with chemoradiotherapy has so far not been adequately investigated. Thus, a benefit-risk ratio for this combination has not yet been established.
Cetuximab in Combination with Platinum-Based Chemotherapy in Recurrent and/or Metastatic Disease: EMR 62 202-002: This randomised study in patients with recurrent and/or metastatic squamous cell cancer of the head and neck who had not received prior chemotherapy for this disease compared the combination of cetuximab and cisplatin or carboplatin plus infusional 5-fluorouracil (222 patients) to the same chemotherapy alone (220 patients). Treatment in the cetuximab arm consisted of up to 6 cycles of platinum-based chemotherapy in combination with cetuximab followed by cetuximab as maintenance therapy until disease progression.
The efficacy data generated in this study are summarised in Table 5.
Click on icon to see table/diagram/image
Patients with a good prognosis as indicated by tumour stage, Karnofsky performance status (KPS) and age had a more pronounced benefit, when cetuximab was added to platinum-based chemotherapy. In contrast to progression free survival time, no benefit in overall survival time could be demonstrated in patients with KPS ≤80 who were ≤65 years.
Pharmacokinetics: Cetuximab pharmacokinetics were studied when cetuximab was administered as monotherapy or in combination with concomitant chemotherapy or radiation therapy in clinical studies. Intravenous infusions of cetuximab exhibited dose-dependent pharmacokinetics at weekly doses ranging from 5-500 mg/m
2 body surface area.
When cetuximab was administered at an initial dose of 400 mg/m
2 body surface area, the mean volume of distribution was approximately equivalent to the vascular space (2.9 L/m
2 with a range of 1.5-6.2 L/m
2). The mean C
max (±standard deviation) was 185±55 mcg/mL. The mean clearance was 0.022 L/hr/m
2 body surface area.
Cetuximab has a long elimination half-life with values ranging from 70-100 hrs at the target dose.
Cetuximab serum concentrations reached stable levels after 3 weeks of cetuximab monotherapy. Mean peak cetuximab concentrations were 155.8 mcg/mL in week 3 and 151.6 mcg/mL in week 8, whereas the corresponding mean trough concentrations were 41.3 mcg/mL and 55.4 mcg/mL, respectively. In a study of cetuximab administered in combination with irinotecan, the mean cetuximab trough levels were 50 mcg/mL in week 12 and 49.4 mcg/mL in week 36.
Several pathways have been described that may contribute to the metabolism of antibodies. All of these pathways involve the biodegradation of the antibody to smaller molecules eg, small peptides or amino acids.
Special Populations: An integrated analysis across all clinical studies showed that the pharmacokinetic characteristics of cetuximab are not influenced by race, age, gender, renal or hepatic status.
Only patients with adequate renal and hepatic function have been investigated to date (serum creatinine ≤1.5-fold, transaminases ≤5-fold and bilirubin ≤1.5-fold the upper limit of normal).
Toxicology: Preclinical Safety Data: Starting at dose levels equivalent to those used in humans, dose-dependent skin alterations were the major findings observed in toxicity studies with cynomolgus monkeys (a chronic repeat-dose toxicity study and an embryo-foetal development study).
An embryo-foetal toxicity study in cynomolgus monkeys revealed no signs of teratogenicity. However, dependent on the dose, an increased incidence of abortion was observed.
Nonclinical data on genotoxicity and local tolerance including accidental administration by routes other than the intended infusion revealed no special hazard for humans.
No formal animal studies have been performed to establish the carcinogenic potential of cetuximab or to determine its effects on male and female fertility.
Toxicity studies with co-administration of cetuximab and chemotherapeutic agents have not been performed.
No nonclinical data on the effect of cetuximab on wound healing are available to date. However, in preclinical wound healing models, EGFR-selective tyrosine kinase inhibitors were shown to retard wound healing.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image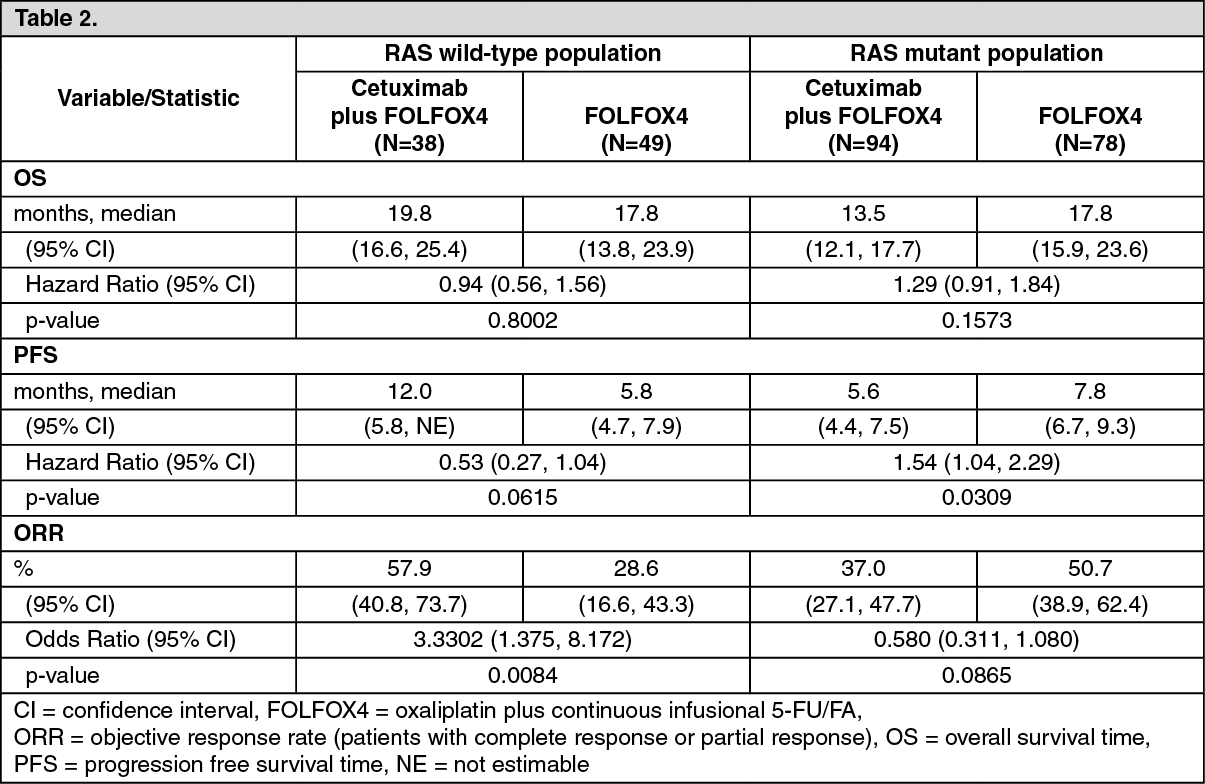 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image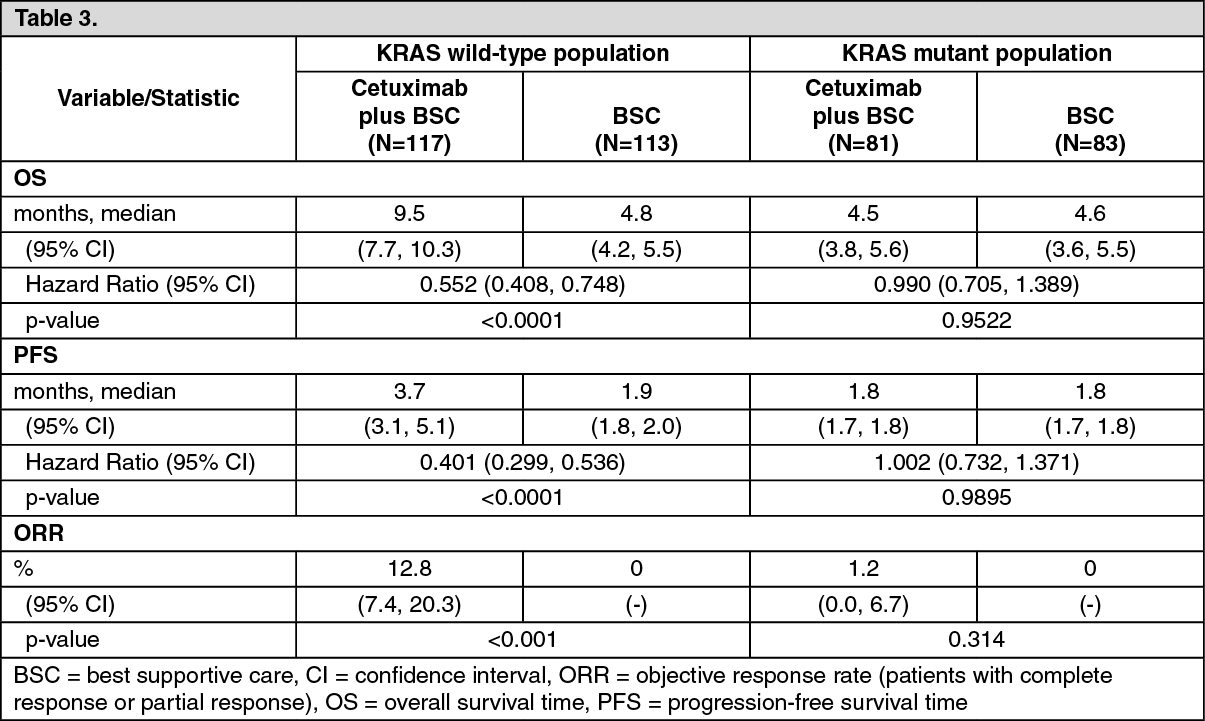 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
