Pharmacology: Mechanism of Action: Alectinib is a tyrosine kinase inhibitor that targets ALK and RET. In nonclinical studies, alectinib inhibited ALK phosphorylation and ALK-mediated activation of the downstream signaling proteins STAT3 and AKT, and decreased tumor cell viability in multiple cell lines harboring ALK fusions, amplifications, or activating mutations. The major active metabolite of alectinib, M4, showed similar in vitro potency and activity.
Alectinib and M4 demonstrated in vitro and in vivo activity against multiple mutant forms of the ALK enzyme, including some mutations identified in NSCLC tumors in patients who have progressed on crizotinib.
In mouse models implanted with tumors carrying ALK fusions, administration of alectinib resulted in antitumor activity and prolonged survival, including in mouse models implanted intracranially with ALK-driven tumor cell lines.
Pharmacodynamics: Cardiac Electrophysiology: The ability of alectinib to prolong the QT interval was assessed in 221 patients administered ALECENSA 600 mg twice daily in clinical studies. ALECENSA did not prolong the QTc (QT corrected for heart rate) interval to any clinically relevant extent. One patient had a maximum post-baseline QTcF value of greater than 500 msec, and one patient had a maximum QTcF change from baseline of greater than 60 msec.
Clinical Studies: Previously Untreated ALK-Positive Metastatic NSCLC: The efficacy of ALECENSA for the treatment of patients with ALK-positive NSCLC who had not received prior systemic therapy for metastatic disease was established in an open-label, randomized, active-controlled, multicenter study (ALEX: NCT02075840). Patients were required to have an ECOG performance status of 0-2 and ALK-positive NSCLC as identified by the VENTANA ALK (D5F3) CDx assay. Neurologically stable patients with treated or untreated central nervous system (CNS) metastases, including leptomeningeal metastases, were eligible; patients with neurologic signs and symptoms due to CNS metastases were required to have completed whole brain radiation or gamma knife irradiation at least 14 days prior to enrollment and be clinically stable. Patients with a baseline QTc > 470 ms were ineligible.
Patients were randomized 1:1 to receive ALECENSA 600 mg orally twice daily or crizotinib 250 mg orally twice daily. Randomization was stratified by ECOG performance status (0/1 vs. 2), race (Asian vs. non-Asian), and the presence or absence of CNS metastases at baseline. Treatment on both arms was continued until disease progression or unacceptable toxicity. The major efficacy outcome measure was progression-free survival (PFS) as determined by investigator assessment according to RECIST v1.1. Additional efficacy outcome measures were PFS as determined by independent review committee (IRC), time to CNS progression by IRC based on RECIST v1.1, objective response rate (ORR) and duration of response (DOR), and overall survival (OS). Additional exploratory outcome measures were CNS objective response rate (CNS-ORR) and CNS duration of response (CNS-DOR) by IRC in patients with CNS metastases at baseline.
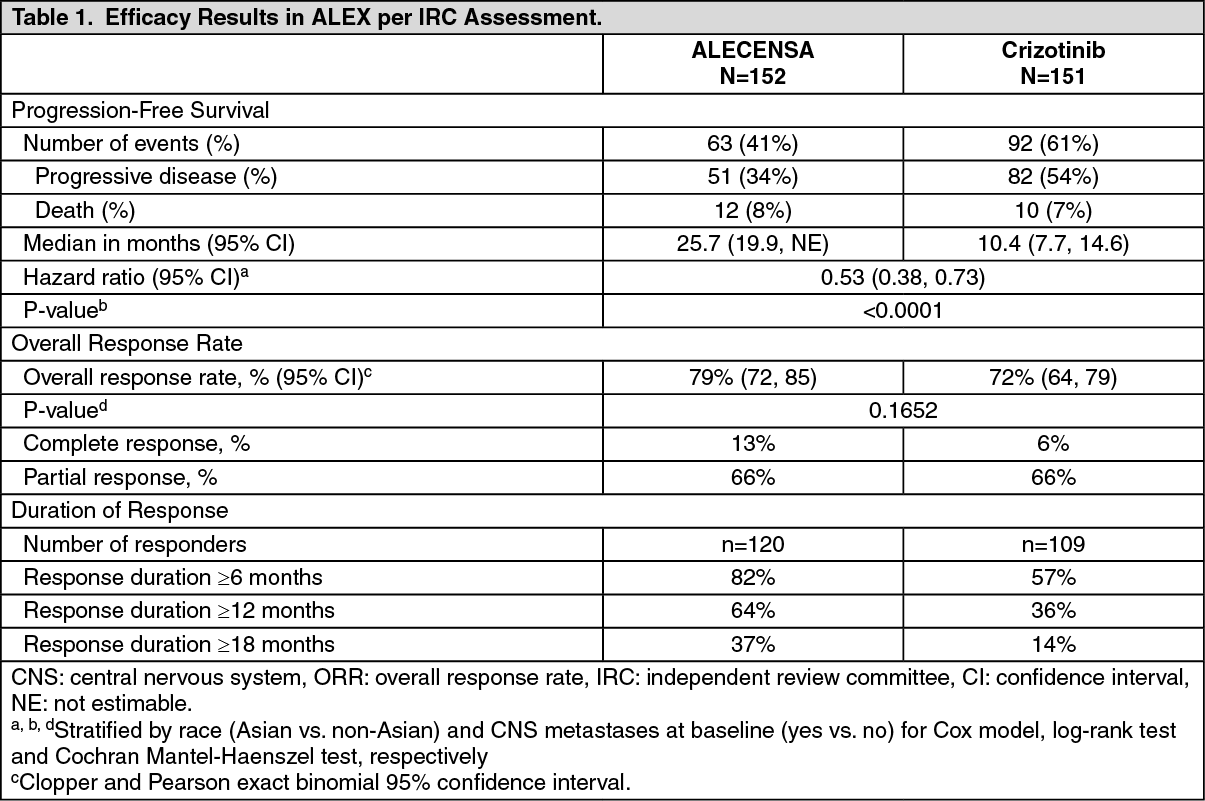
A total of 303 patients were randomized to ALECENSA (n=152) or crizotinib (n=151). The demographic characteristics of the study population were 56% female, median age 56 years (range: 18 to 91 years), 50% White, 46% Asian, 1% Black, and 3% other races. The majority of patients had adenocarcinoma (92%) and never smoked (63%). CNS metastases were present in 40% (n=122) of patients: of these, 43 patients had measurable CNS lesions as determined by an IRC. The ALEX study demonstrated a significant improvement in PFS. The time to cause-specific CNS progression as assessed by IRC was also significantly improved; there was a lower incidence of progression in the CNS as the first site of disease progression, alone or with concurrent systemic progression, in the ALECENSA arm (12%) as compared to the crizotinib arm (45%). Efficacy results from ALEX are summarized in Table 1 and figure. (See Table 1 and figure.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Results for PFS as determined by investigator assessment (HR=0.48 [95% CI: 0.35-0.66], stratified log-rank p<0.0001) were similar to that observed by IRC. At the data cutoff point overall survival data was not mature.
The results of prespecified exploratory analyses of CNS response rate in patients with measurable CNS lesions at baseline are summarized in Table 2. (See Table 2.)
Click on icon to see table/diagram/image
ALK-Positive Metastatic NSCLC Previously Treated with Crizotinib: The safety and efficacy of ALECENSA were established in two single-arm, multicenter clinical trials NP28761: (NCT01588028) and NP28673 (NCT01801111). Patients with locally advanced or metastatic ALK-positive NSCLC, who have progressed on crizotinib, with documented ALK-positive NSCLC based on an FDA-approved test, and ECOG PS of 0-2 were enrolled in both studies. Eligibility criteria permitted enrollment of patients with prior chemotherapy and prior CNS radiotherapy provided that CNS metastases were stable for at least two weeks. All patients received ALECENSA 600 mg orally twice daily. The major efficacy outcome measure in both studies was objective response rate (ORR) according to Response Evaluation Criteria in Solid Tumours (RECIST v1.1) as evaluated per Independent Review Committee (IRC). Additional outcome measures as evaluated by the IRC included duration of response (DOR), CNS ORR, and CNS DOR.
NP28761 was conducted in North America and enrolled 87 patients. Baseline demographic and disease characteristics were median age 54 years old (range 29 to 79, 18% 65 and over), 84% White and 8% Asian, 55% female, 35% ECOG PS 0 and 55% ECOG PS 1, 100% never or former smokers, 99% Stage IV, 94% adenocarcinoma, and 74% prior chemotherapy. The most common sites of extra-thoracic metastasis included 60% CNS (of whom 65% had received CNS radiation), 43% lymph nodes, 36% bone, and 34% liver.
NP28673 was conducted internationally and enrolled 138 patients. Baseline demographic and disease characteristics in NP28673 were median age 52 years old (range 22 to 79, 10% 65 and over), 67% White and 26% Asian, 56% female, 32% ECOG PS 0 and 59% ECOG PS 1, 98% never or former smokers, 99% Stage IV, 96% adenocarcinoma, and 80% prior chemotherapy. The most common sites of extra-thoracic metastasis included 61% CNS (of whom 73% had received CNS radiation), 51% bone, 38% lymph nodes, and 30% liver.
Efficacy results from NP28761 and NP28673 in all treated patients are summarized in Table 3. The median duration of follow-up on Study NP28761 was 4.8 months for both IRC and Investigator assessments and on Study NP28673, 10.9 months for IRC assessment and 7.0 months for Investigator assessment. All responses were partial responses. (See Table 3.)
Click on icon to see table/diagram/image
An assessment of ORR and duration of response for CNS metastases in the subgroup of 51 patients in NP28761 and NP28673 with baseline measurable lesions in the CNS according to RECIST v1.1 are summarized in Table 4. Thirty-five (69%) patients with measurable CNS lesions had received prior brain radiation, including 25 (49%) who completed radiation treatment at least 6 months before starting treatment with ALECENSA. Responses were observed irrespective of prior brain radiation status. (See Table 4.)
Click on icon to see table/diagram/image
Pharmacokinetics: The pharmacokinetics of alectinib and its major active metabolite M4 have been characterized in patients with ALK-positive NSCLC and healthy subjects.
In patients with ALK-positive NSCLC, the geometric mean (coefficient of variation %) steady-state maximal concentration (C
max,ss) for alectinib was 665 ng/mL (44%) and for M4 was 246 ng/mL (45%) with peak to trough concentration ratio of 1.2. The geometric mean steady-state area under the curve from 0 to 12 hours (AUC
0-12h,ss) for alectinib was 7,430 ng*h/mL (46%) and for M4 was 2,810 ng*h/mL (46%). Alectinib exposure is dose proportional across the dose range of 460 mg to 900 mg (i.e., 0.75 to 1.5 times the approved recommended dosage) under fed conditions. Alectinib and M4 reached steady-state concentrations by day 7. The geometric mean accumulation was approximately 6-fold for both alectinib and M4.
Absorption: Alectinib reached maximal concentrations at 4 hours following administration of ALECENSA 600 mg twice daily under fed conditions in patients with ALK-positive NSCLC.
The absolute bioavailability of alectinib was 37% (90% CI: 34%, 40%) under fed conditions.
A high-fat, high-calorie meal increased the combined exposure (AUC
0-inf) of alectinib plus M4 by 3.1-fold (90% CI: 2.7, 3.6) following oral administration of a single 600 mg dose of ALECENSA.
Distribution: The apparent volume of distribution is 4,016 L for alectinib and 10,093 L for M4.
Alectinib and M4 are bound to human plasma proteins greater than 99%, independent of drug concentration.
Alectinib concentrations in the cerebrospinal fluid in patients with ALK-positive NSCLC approximate estimated alectinib free concentrations in the plasma.
In vitro studies suggest that alectinib is not a substrate of P-glycoprotein (P-gp), but M4 is a substrate of P-gp. Alectinib and M4 are not substrates of breast cancer resistance protein (BCRP), organic anion-transporting polypeptide (OATP) 1B1, or OATP1B3.
Elimination: The apparent clearance (CL/F) is 81.9 L/hour for alectinib and 217 L/hour for M4. The geometric mean elimination half-life is 33 hours for alectinib and 31 hours for M4 in patients with ALK-positive NSCLC.
Metabolism: Alectinib is metabolized by CYP3A4 to its major active metabolite M4. The geometric mean metabolite/parent exposure ratio at steady-state is 0.40. M4 is subsequently metabolized by CYP3A4. Alectinib and M4 were the main circulating moieties in plasma, constituting 76% of the total radioactivity.
Excretion: Ninety-eight percent of the radioactivity was excreted in feces following oral administration of a single radiolabeled dose of alectinib, under fed conditions. Eighty-four percent of the dose was excreted in the feces as unchanged alectinib and 6% of the dose was excreted as M4. Excretion of radioactivity in urine was less than 0.5% of administered radiolabeled dose of alectinib.
Specific Populations: Age (21 to 83 years), body weight (38 to 128 kg), mild hepatic impairment (total bilirubin ≤ ULN and AST > ULN or total bilirubin 1 to ≤ 1.5 × ULN and AST any value), mild to moderate renal impairment (creatinine clearance 30 to 89 mL/min), race (White, Asian, and Other), and sex had no clinically meaningful effect on the systemic exposure of alectinib and M4. The pharmacokinetics of alectinib have not been studied in patients with severe renal impairment (creatinine clearance < 30 mL/min), or end-stage renal disease.
Hepatic Impairment: Following administration of a single oral dose of 300 mg ALECENSA, the geometric mean ratio [90% confidence interval] for the combined AUC
inf of alectinib and M4 in subjects with moderate hepatic impairment (Child-Pugh B) was 1.36 [0.947, 1.96] and in subjects with severe hepatic impairment (Child-Pugh C) was 1.76 [0.984, 3.15] as compared to that in subjects with normal hepatic function. The combined C
max of alectinib and M4 was comparable among the three groups. No dose adjustment is recommended for patients with mild or moderate hepatic impairment. The recommended dose of ALECENSA in patients with severe hepatic impairment is 450 mg orally twice daily [see Dosage & Administration and Hepatic Impairment under Precautions].
Drug Interactions: Effect of Other Drugs on Alectinib: No clinically meaningful effect on the combined exposure of alectinib plus M4 was observed in clinical studies following co-administration of ALECENSA with a strong CYP3A inhibitor (posaconazole), a strong CYP3A inducer (rifampin), or an acid-reducing agent (esomeprazole).
Effect of Alectinib on Other Drugs: No clinically meaningful effect on the exposure of midazolam (sensitive CYP3A substrate) or repaglinide (sensitive CYP2C8 substrate) is expected following co-administration with ALECENSA.
In vitro studies suggest that alectinib and M4 do not inhibit CYP1A2, 2B6, 2C9, 2C19 or 2D6.
In vitro studies suggest that alectinib and M4 inhibit P-gp and BCRP. Alectinib did not inhibit OATP1B1, OATP1B3, OAT1, OAT3, or OCT2 transport activity in vitro.
Toxicology: Nonclinical Toxicology: Carcinogenesis, Mutagenesis, Impairment of Fertility: Carcinogenicity studies with alectinib have not been conducted.
Alectinib was not mutagenic in vitro in the bacterial reverse mutation (Ames) assay, but was positive with an increased number of micronuclei in a rat bone marrow micronucleus test. The mechanism of micronucleus induction was abnormal chromosome segregation (aneugenicity) and not a clastogenic effect on chromosomes.
No studies in animals have been performed to evaluate the effect of alectinib on fertility. No adverse effects on male and female reproductive organs were observed in general toxicology studies conducted in rats and monkeys.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image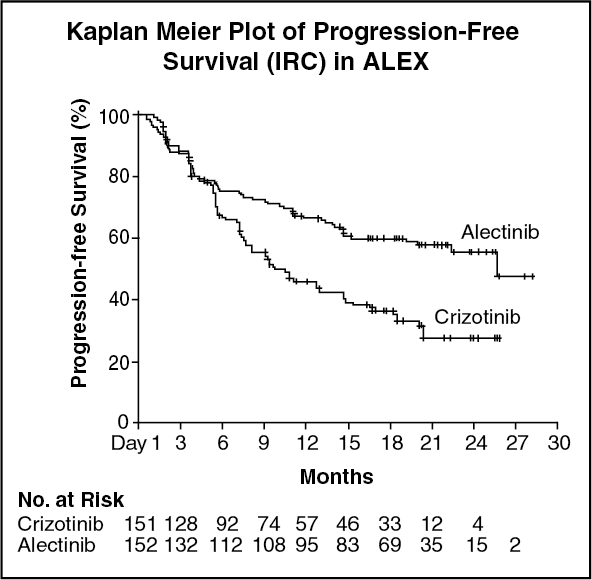 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image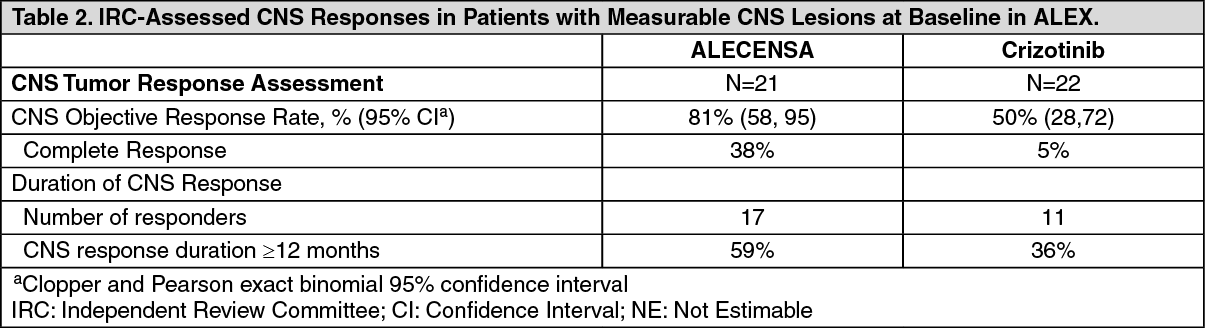 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image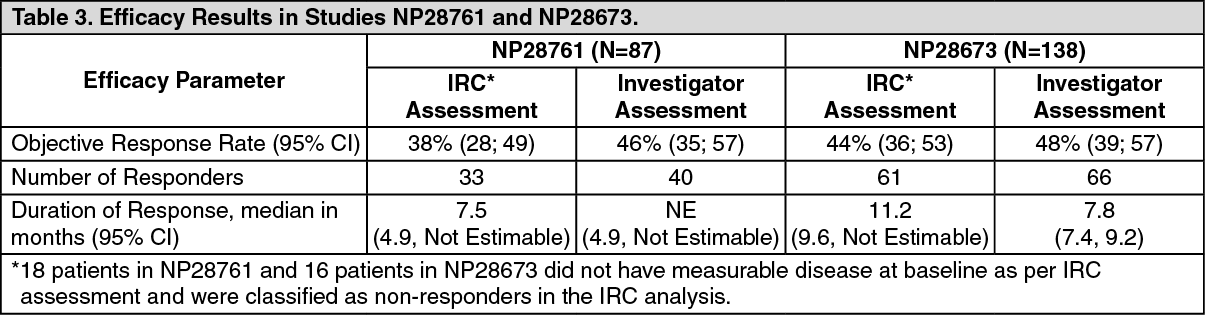 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image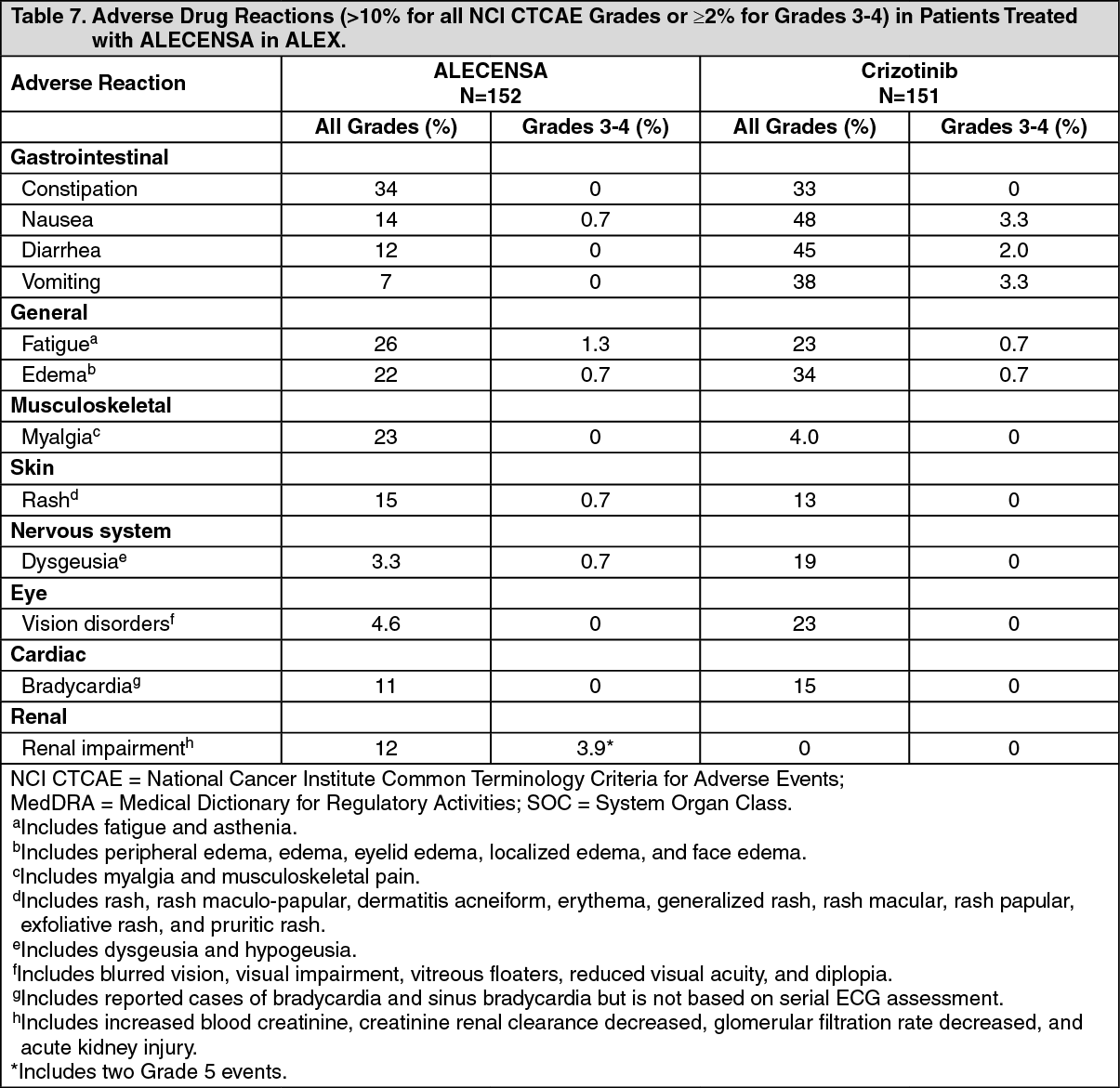 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image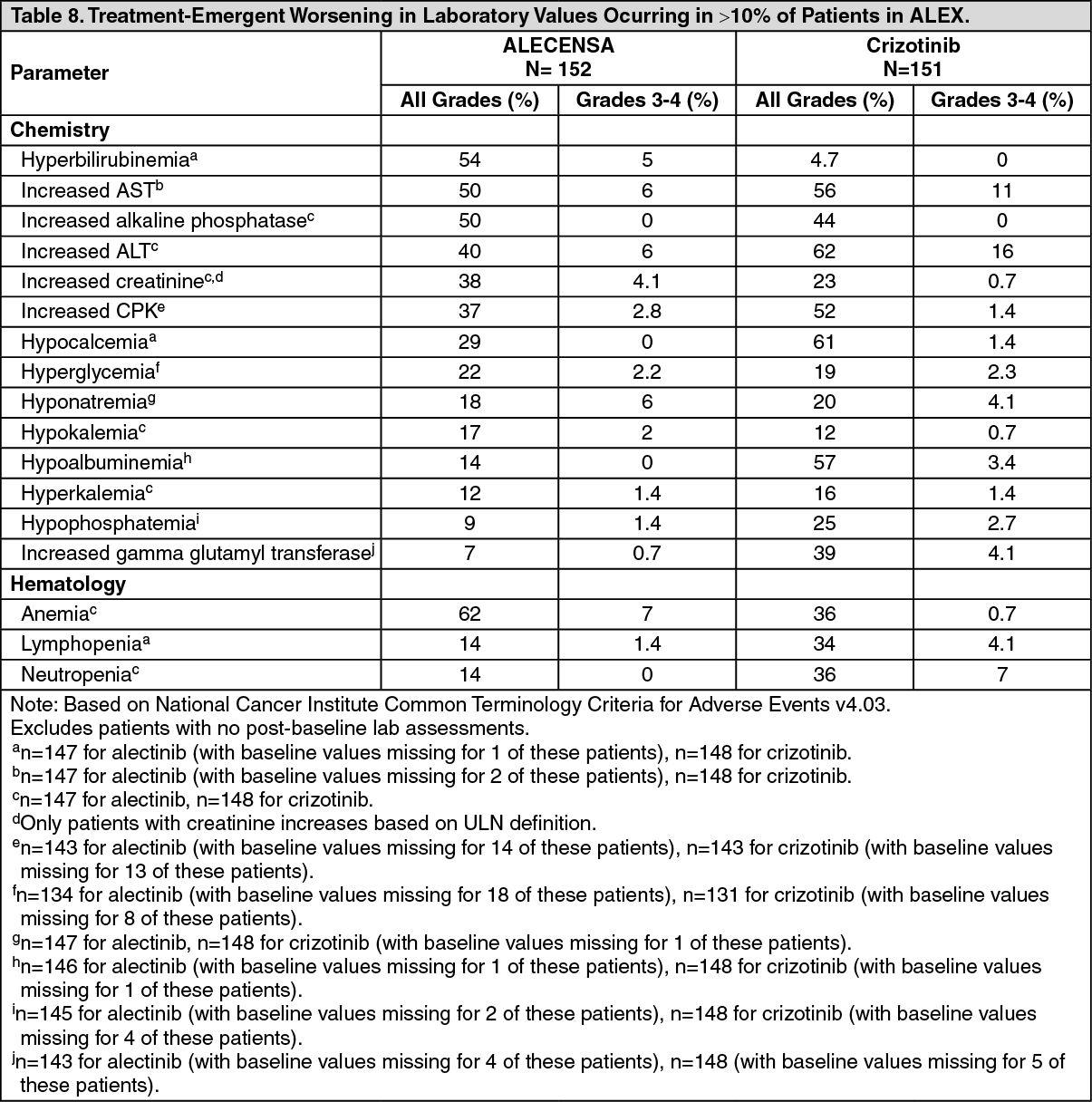 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image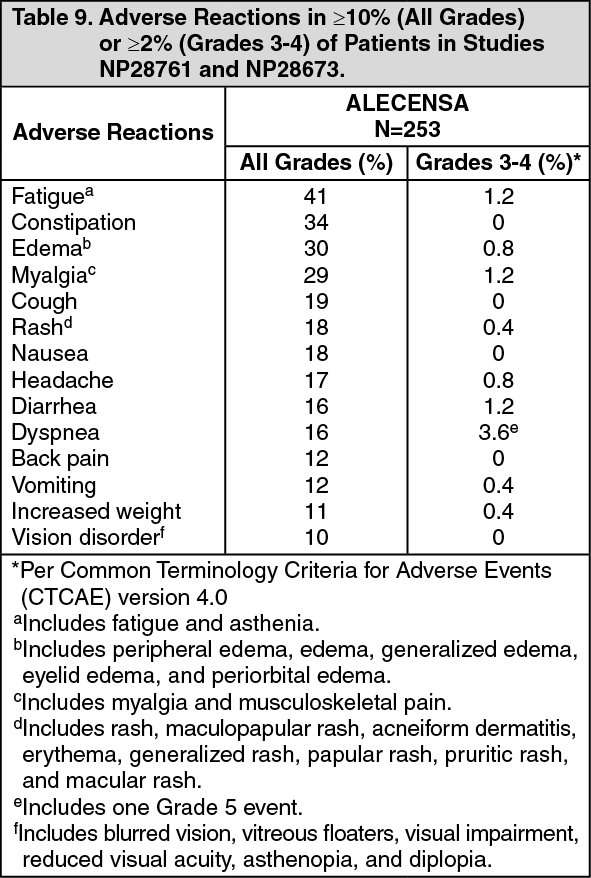 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
