


 Sign Out
Sign Out

Clinical Trials Experience: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Previously Untreated ALK-Positive Metastatic NSCLC: The safety of ALECENSA was evaluated in 152 patients with ALK-positive NSCLC in the ALEX study. The median duration of exposure to ALECENSA was 17.9 months. Patient characteristics of the ALEX study population (n=303) were: median age 56 years, age less than 65 (77%), female (56%), Caucasian (50%), Asian (46%), adenocarcinoma histology (92%), never smoker (63%), and ECOG PS 0 or 1 (93%).
Serious adverse reactions occurred in 28% of patients treated with ALECENSA; serious adverse reactions reported in 2% or more of patients treated with ALECENSA were pneumonia (4.6%), and renal impairment (3.9%). Grade ≥3 adverse events were reported for 41% of patients in the ALECENSA arm. Fatal adverse reactions occurred in 3.3% of patients treated with ALECENSA; these were renal impairment (2 patients), sudden death, cardiac arrest, and pneumonia (1 patient each). Permanent discontinuation of ALECENSA for adverse reactions occurred in 11% of patients. Adverse drug reactions that led to discontinuation of ALECENSA in 1% or more of patients were renal impairment (2.0%), hyperbilirubinemia (1.3%), increased ALT (1.3%), and increased AST (1.3%). Dose reductions and drug interruption due to adverse reactions occurred in 16% and 19% of patients, respectively, in the ALECENSA arm. The most frequent adverse reactions that led to dose modifications in the ALECENSA arm were hyperbilirubinemia (6%), increased AST (5%), increased ALT (4.6%), and pneumonia (3.3%).
Tables 7 and 8 summarize the common adverse reactions and laboratory abnormalities observed in ALEX. (See Table 7.)
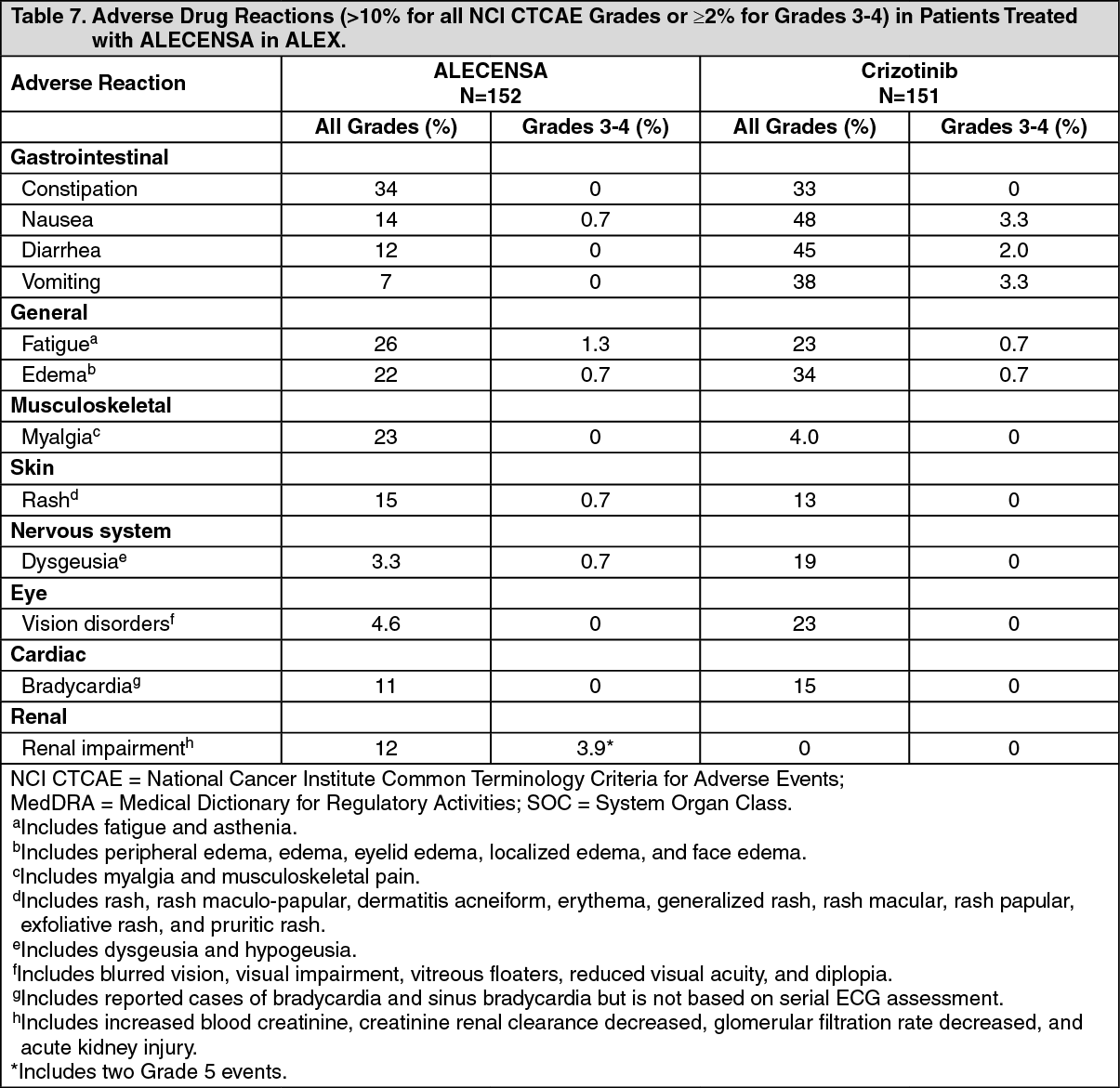 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe following additional clinically significant adverse drug reactions were observed in patients treated with ALECENSA: weight gain (9.9%), photosensitivity reaction (5.3%), stomatitis (3.3%), interstitial lung disease (1.3%), and drug-induced liver injury (1.3%). (See Table 8.)
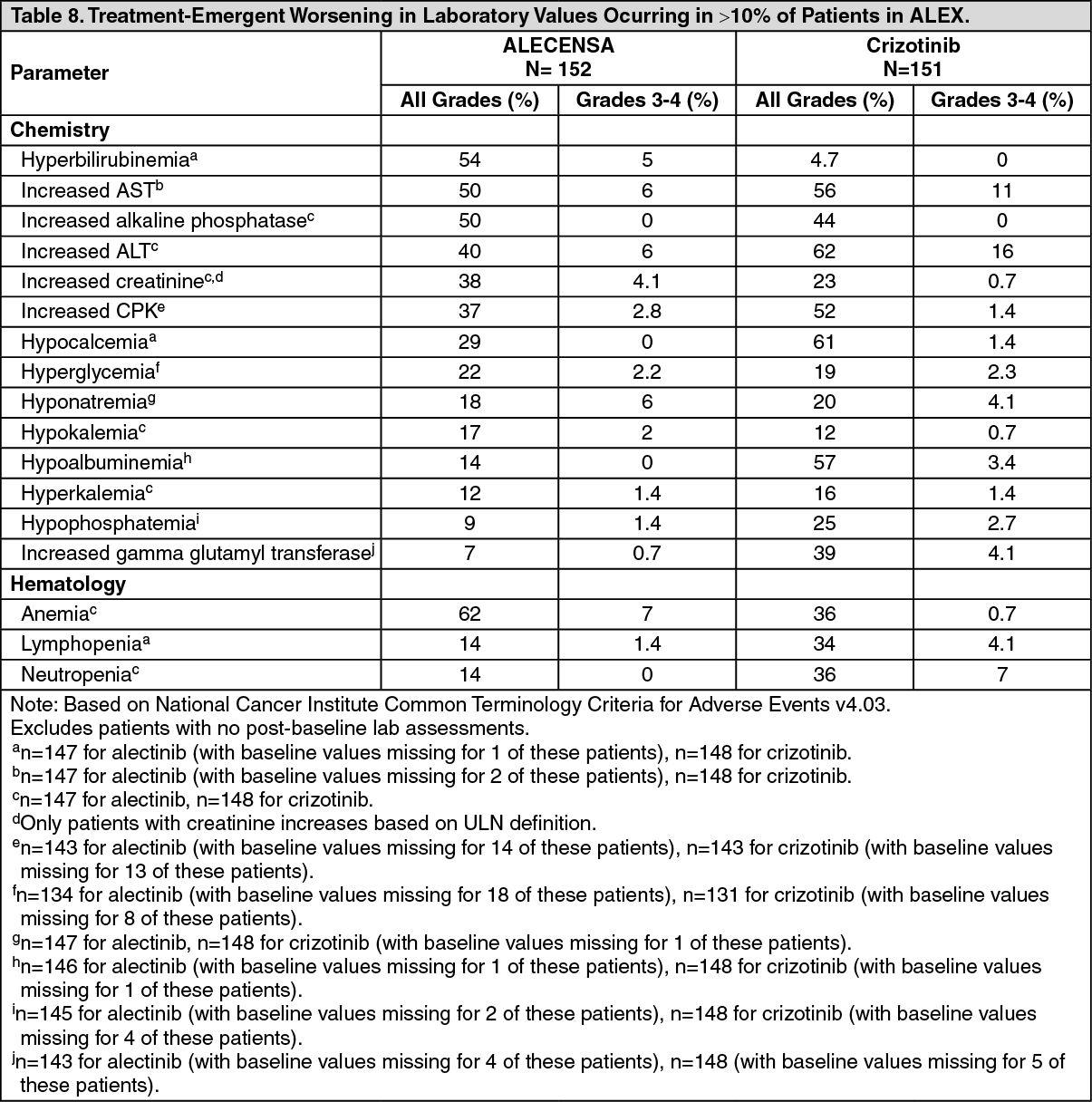 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageALK-Positive Metastatic NSCLC Previously Treated with Crizotinib: The safety of ALECENSA was evaluated in 253 patients with ALK-positive non-small cell lung cancer (NSCLC) treated with ALECENSA in two clinical trials, Studies NP28761 and NP28673. The median duration of exposure to ALECENSA was 9.3 months. One hundred sixty-nine patients (67%) were exposed to ALECENSA for more than 6 months, and 100 patients (40%) for more than one year. The population characteristics were: median age 53 years, age less than 65 (86%), female (55%), White (74%), Asian (18%), NSCLC adenocarcinoma histology (96%), never or former smoker (98%), ECOG Performance Status (PS) 0 or 1 (91%), and prior chemotherapy treatment (78%).
Serious adverse reactions occurred in 19% of patients; the most frequently reported serious adverse reactions were pulmonary embolism (1.2%), dyspnea (1.2%), and hyperbilirubinemia (1.2%). Fatal adverse reactions occurred in 2.8% of patients and included hemorrhage (0.8%), intestinal perforation (0.4%), dyspnea (0.4%), pulmonary embolism (0.4%), and endocarditis (0.4%). Permanent discontinuation of ALECENSA for adverse reactions occurred in 6% of patients. The most frequent adverse reactions that led to permanent discontinuation were hyperbilirubinemia (1.6%), increased ALT levels (1.6%), and increased AST levels (1.2%). Overall, 23% of patients initiating treatment at the recommended dose required at least one dose reduction. The median time to first dose reduction was 48 days. The most frequent adverse reactions that led to dose reductions or interruptions were elevations in bilirubin (6%), CPK (4.3%), ALT (4.0%), and AST (2.8%), and vomiting (2.8%).
Tables 9 and 10 summarize the common adverse reactions and laboratory abnormalities observed in Studies NP28761 and NP28673. (See Table 9.)
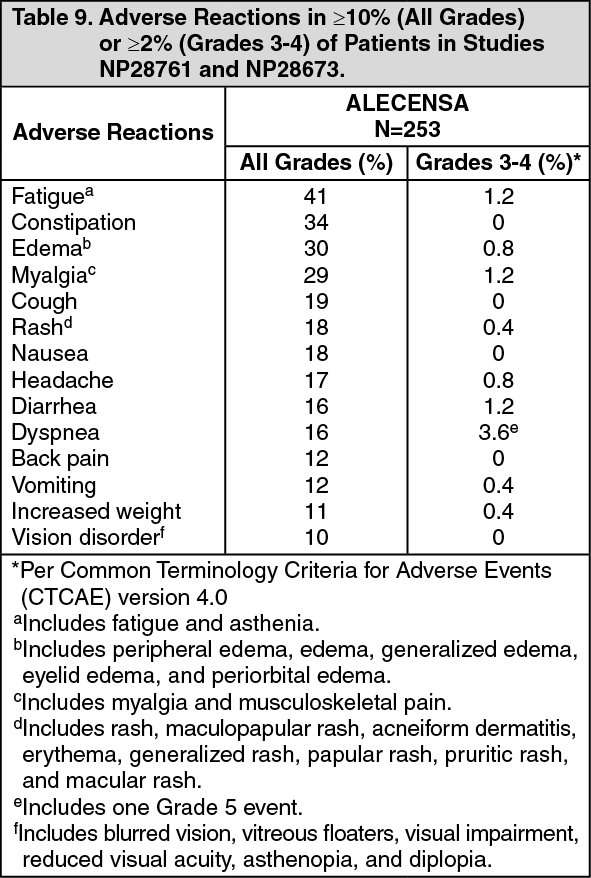 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAn additional clinically significant adverse drug reaction was photosensitivity, which occurred in 9.9% of patients exposed to ALECENSA in Studies NP28761 and NP28673. Patients were advised to avoid sun exposure and to use broad-spectrum sunscreen. The incidence of Grade 2 photosensitivity was 0.4%; the remaining events were Grade 1 in severity. (See Table 10.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePostmarketing Experience: The following adverse reactions have been identified during post approval use of ALECENSA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and lymphatic system disorders: hemolytic anemia.
View ADR Monitoring Form