


 Sign Out
Sign Out


Inhibitory effect on DPP-4 and suppressive action on GLP-1 degradation: Teneligliptin inhibited the activity of DPP-4 in human plasma in a concentration-dependent manner, with IC50 of 1.75 nmol/L (in vitro).
Teneligliptin prevented the degradation of active GLP-1 in rat plasma in a concentration-dependent manner (in vitro).
In a glucose tolerance test in Zucker Fatty rats, a model of obesity with insulin resistance and impaired glucose tolerance, a single oral administration of teneligliptin increased plasma active GLP-1 and plasma insulin levels.
In patients with type 2 diabetes mellitus, once-daily administration of teneligliptin 20 mg inhibited plasma DPP-4 activity and increased the concentration of active GLP-1 in plasma.
Improvement of glucose tolerance: In a glucose tolerance test in Zucker Fatty rats, a model of obesity with insulin resistance and impaired glucose tolerance, a single oral administration of teneligliptin improved post-loaded hyperglycemia.
In patients with type 2 diabetes mellitus, once-daily administration of teneligliptin 20 mg improved blood glucose after breakfast, lunch and dinner and fasting blood glucose.
Clinical efficacy: Monotherapy: Placebo-controlled double-blind comparative study (dose-finding study): Patients with type 2 diabetes mellitus inadequately controlled on diet and exercise therapy received 10 mg, 20 mg or 40 mg of teneligliptin or placebo once daily for 12 weeks. The difference (Least mean square value [95% confidence interval]) from the placebo group of the change in HbA1c level from baseline at Week 12 was -0.90 [-1.06, -0.75]% in the 20 mg administration group (n = 79), and -1.01 [-1.16, -0.86]% in the 40 mg administration group (n = 81). The incidence of adverse drug reaction of hypoglycaemia was 0% (0/79 patients) in the 20 mg administration group and 3.7% (3/81 patients) in the 40 mg administration group. (The usual dosage of this drug is 20 mg once daily, as teneligliptin, and the maximum approved dosage is 40 mg once daily.)
Placebo-controlled double-blind comparative study (confirmatory study): Patients with type 2 diabetes mellitus inadequately controlled on diet and exercise therapy (n = 203) received 20 mg of teneligliptin or placebo once daily for 12 weeks. The result is shown in the following table. No adverse drug reaction of hypoglycaemia was observed in the teneligliptin-treated group. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageCombination therapy with other hypoglycemic agents: Combination therapy with sulfonylurea: Patients with type 2 diabetes mellitus inadequately controlled on diet and exercise therapy and glimepiride (n = 194) received 20 mg of teneligliptin or placebo once daily for 12 weeks. The result is shown in the following table. (See Table 2.)
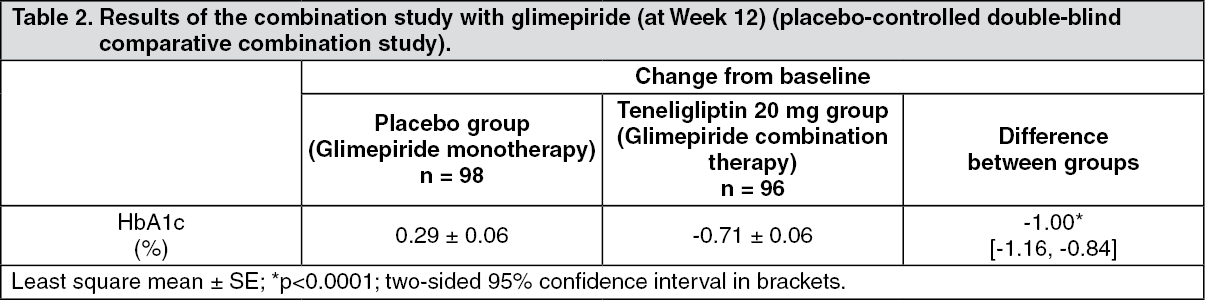 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageWhen patients received the dose of 20 mg or 40 mg (when the dose was increased) once a day with glimepiride after Week 12, the change (mean ± SD) in HbA1c level from baseline at Week 52 was -0.56 ± 0.87% (n = 96). The incidence of adverse drug reaction of hypoglycaemia up to Week 52 was 7.3% (7/96 patients).
Combination therapy with thiazolidine: Patients with type 2 diabetes mellitus inadequately controlled on diet and exercise therapy and pioglitazone (n = 204) received 20 mg of teneligliptin or placebo once daily for 12 weeks. The result is shown in the following table. (See Table 3.)
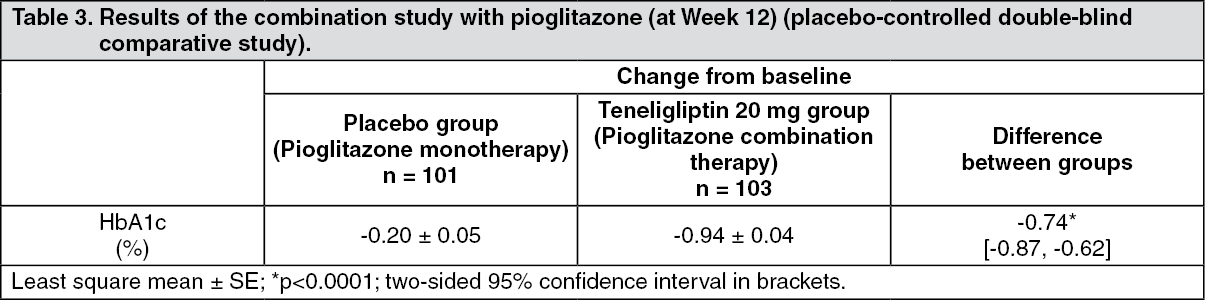 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageWhen patients received the dose of 20 mg or 40 mg (when the dose was increased) once a day with pioglitazone after Week 12, the change (mean ± SD) in HbA1c level from baseline at Week 52 was -0.86 ± 0.66% (n = 103). The incidence of adverse drug reaction of hypoglycaemia up to Week 52 was 1.9% (2/103 patients).
Combination therapy with insulin: Patients with type 2 diabetes mellitus inadequately controlled on insulin monotherapy (mixed type [short-acting or rapid-acting type insulin content 25% or 30%], intermediate type, or long-acting type at a daily dose of 8 units or more but 40 units or less) in addition to diet and exercise therapy (n=148) received 20 mg of teneligliptin or placebo once daily for 16 weeks. The result is shown in the following table. (See Table 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageWhen patients received the dose of 20 mg or 40 mg (when the dose was increased) once a day with insulin after Week 16, the change (mean ± SD) in HbA1c level from baseline at Week 52 was -0.81 ± 0.93% (n = 77). The incidence of adverse drug reaction of hypoglycaemia up to Week 52 was 15.6% (12/77 patients).
Long-term study (monotherapy and combination therapy with glinide, biguanide, or α-glucosidase inhibitor): Patients with type 2 diabetes mellitus inadequately controlled on glinide, biguanide, or an α-glucosidase-inhibitor in addition to dietary and exercise therapy (n = 462) received 20 mg or 40 mg (when the dose was increased) of teneligliptin once daily for 52 weeks. The change in HbA1c level from baseline at Week 52 was -0.63 ± 0.64% (n = 212) in monotherapy, -0.76 ± 0.70% (n = 80) in combination with glinide, -0.78 ± 0.75% (n = 95) in combination with biguanide, and -0.89 ± 0.64% (n = 75) in combination with an α-glucosidase inhibitor. The incidence of adverse drug reactions of hypoglycaemia up to Week 52 was 1.4% (3/212 patients) in monotherapy, 3.8% (3/80 patients) in combination with glinide, 1.1% (1/95 patients) in combination with biguanide, and 1.3% (1/75 patients) in combination with an α-glucosidase inhibitor.
Pharmacokinetics: Plasma concentrations: Single administration: Plasma concentration–time profiles of teneligliptin and its pharmacokinetic parameters after single oral administration of 20 mg and 40 mg of teneligliptin to healthy adults under fasting condition are as shown as follows. (See Figure and Table 5.)
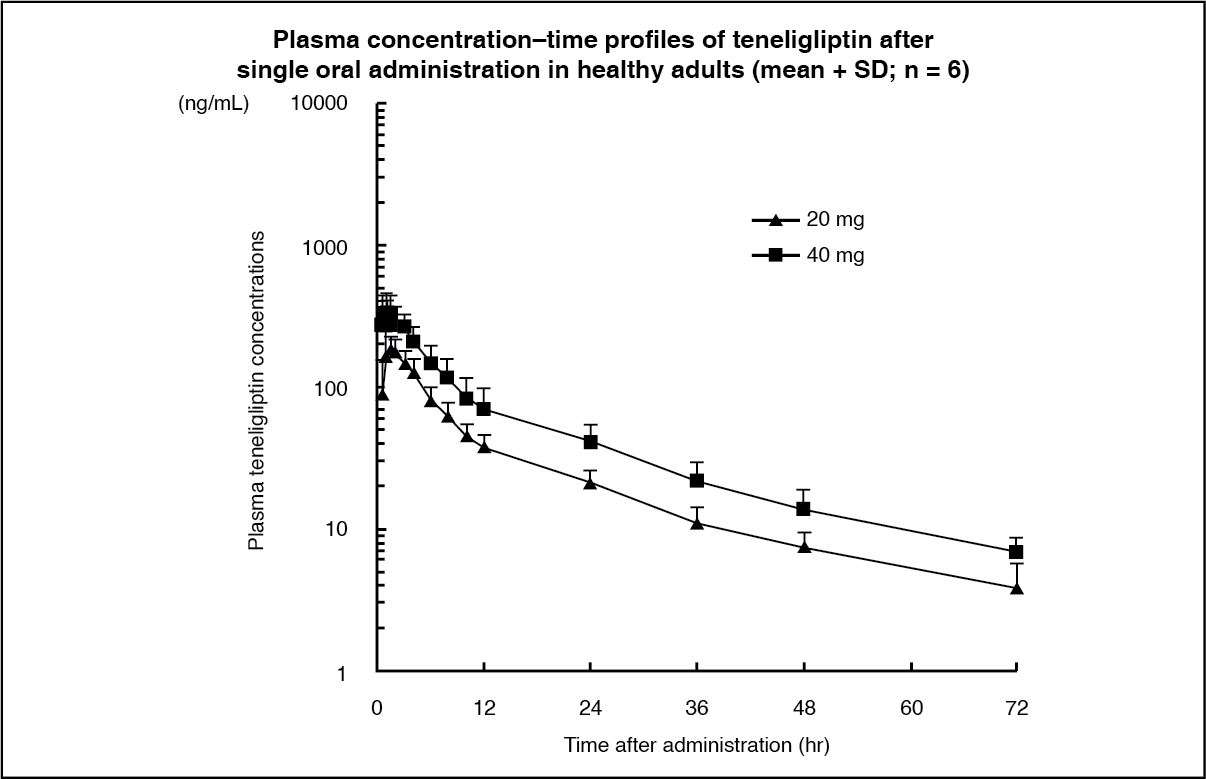 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageRepeated administration: The pharmacokinetic parameters of teneligliptin after repeated oral administration of 20 mg of teneligliptin once daily for 7 days to healthy adults 30 minutes before breakfast are as shown as follows and were estimated to reach steady state within 7 days. (See Table 6.)
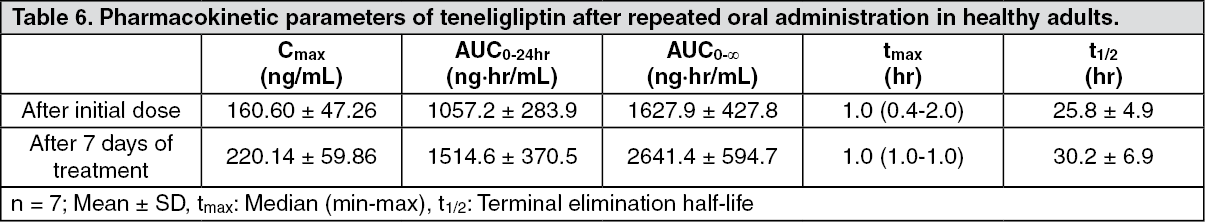 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageEffects of food: When healthy adults were administered a single oral dose of 20 mg of teneligliptin after meals, there was a 20% decrease in Cmax compared to under fasting condition and a prolongation in tmax from 1.1 hours to 2.6 hours, while there was no difference in AUC. (See Table 7.)
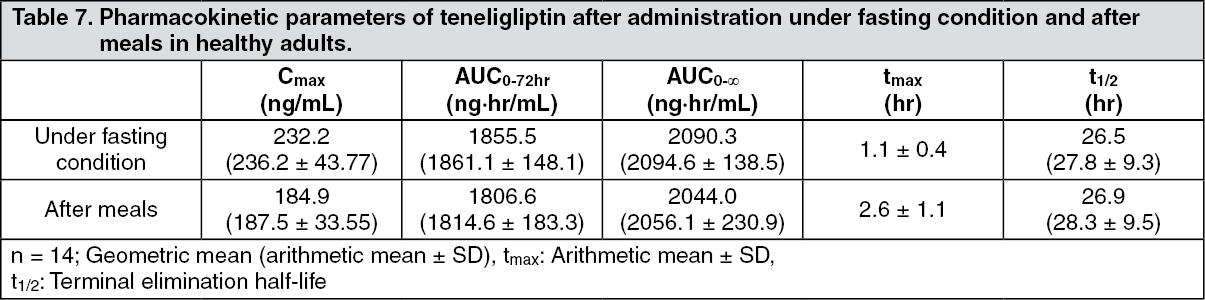 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePlasma protein binding: The in vitro protein bindings of 14C-labeled teneligliptin (20, 100 and 500 ng/mL) to human plasma were 77.6% to 82.2%.
Metabolism: When healthy adults (n = 6) were administered a single oral dose of 14C-labeled teneligliptin 20 mg, the unchanged drug and its metabolites, M1, M2, M3, M4 and M5, were found in plasma. The AUC0-∞ ratios of teneligliptin and its metabolites M1, M2, M3, M4 and M5 to total radioactivity, which were calculated based on plasma radioactive concentrations up to 72 hours after administration, were 71.1%, 14.7%, 1.3%, 1.3%, 0.3% and 1.1%, respectively.
CYP3A4 and flavin-containing monooxygenase (FMO1 and FMO3) are primarily involved in metabolism of teneligliptin. While teneligliptin had weak inhibitory effect on CYP2D6, CYP3A4 and FMO (IC50: 489.4, 197.5 and 467.2 μmol/L, respectively), it had no inhibitory effect on CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C8/9, CYP2C19 and CYP2E1 and did not induce CYP1A2 and CYP3A4 (in vitro).
Excretion: When healthy adults were administered a single oral dose of 20 mg and 40 mg of teneligliptin under fasting condition (n = 6 each), 21.0% to 22.1% of the administered dose was excreted unchanged drug in the urine, and renal clearance was 37 to 39 mL/hr/kg.
When healthy adults (n = 6) were administered a single oral dose of 14C-labeled teneligliptin 20 mg, 45.4% and 46.5% of the administered radioactive dose was excreted in the urine and feces, respectively. Cumulative urinary excretion of unchanged drug, M1, M2 and M3 to the doses up to 120 hours after administration was 14.8%, 17.7%, 1.4% and 1.9%, respectively, and cumulative fecal excretion of unchanged drug, M1, M3, M4 and M5 was 26.1%, 4.0%, 1.6%, 0.3% and 1.3%, respectively.
Teneligliptin is a substrate of P-glycoprotein and inhibited digoxin transport mediated by P-glycoprotein to 42.5% at a concentration of 99 μmol/L. In addition, while teneligliptin had weak inhibitory effect on organic anion transporter (OAT) 3 expressed in the kidney (IC50: 99.2 μmol/L), it had no inhibitory effect on OAT 1, organic cation transporter (OCT) 2, organic anion-transporting polypeptide (OATP) 1B1 and OATP 1B3 (in vitro).
Subjects with renal impairment: When subjects with renal impairment were administered a single oral dose of 20 mg of teneligliptin, there was no marked change in Cmax and t½ of teneligliptin according to the severity of renal impairment. On the other hand, AUC0-∞ in subjects with mild (50≤Ccr≤80 mL/min), moderate (30≤Ccr<50 mL/min) and severe (Ccr<30 mL/min) renal impairment was approximately 1.25 times, 1.68 times and 1.49 times, respectively, compared to that in healthy adults, and AUC0-43hr in subjects with end stage renal failure was approximately 1.16 times compared to that in healthy adults. In addition, 15.6% of the administered teneligliptin dose was eliminated by haemodialysis. (See Table 8.)
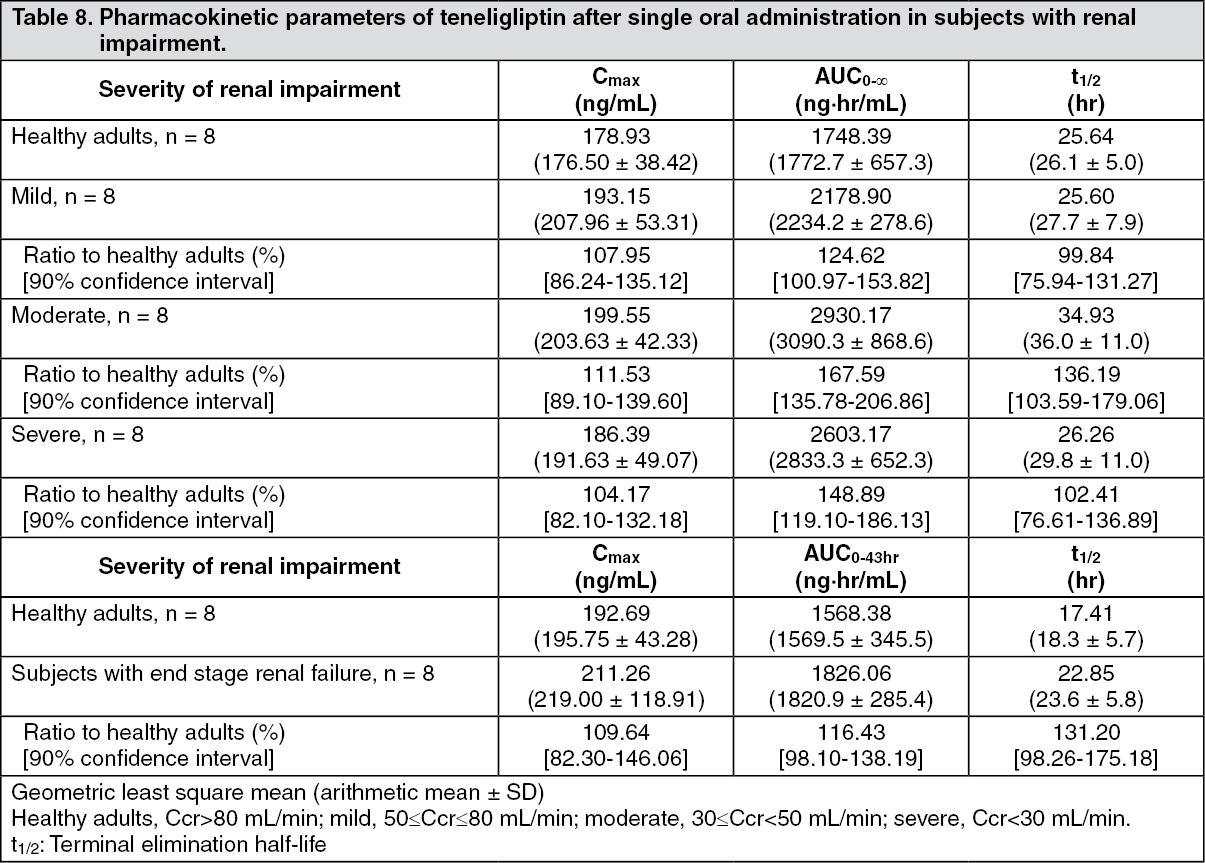 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSubjects with hepatic impairment: When subjects with hepatic impairment were administered a single oral dose of 20 mg of teneligliptin, Cmax of teneligliptin in subjects with mild (total score of 5 to 6 on the Child-Pugh Classification) and moderate (total score of 7 to 9 on the Child-Pugh Classification) hepatic impairment was approximately 1.25 times and 1.38 times, respectively, compared to that in healthy adults, and AUC0-∞ was approximately 1.46 times and 1.59 times, respectively. There has been no clinical experience in subjects with severe hepatic impairment (total score of >9 on the Child-Pugh Classification). (See Table 9.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePharmacokinetics in the elderly: When healthy elderly subjects (aged ≥65 years and ≤75 years, n = 12) and non-elderly subjects (aged ≥45 years and <65 years, n = 12) were administered a single oral dose of 20 mg of teneligliptin under fasting condition, the ratio of geometric least square means (90% confidence interval) of Cmax, AUC0-∞ and t½ in elderly subjects to those in non-elderly subjects was 1.006 (0.871-1.163), 1.090 (0.975-1.218) and 1.054 (0.911-1.219), and the parameters of teneligliptin were similar in elderly and non-elderly subjects.
Drug interactions: Coadministration with ketoconazole: The influence of concomitant administration of ketoconazole on the pharmacokinetics of teneligliptin is shown in the following table. (See Table 10.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageCoadministration with other antidiabetic drugs: When teneligliptin was concomitantly administered with canagliflozin, pioglitazone, glimepiride or metformin, no obvious effect of concomitant administration on the pharmacokinetics of teneligliptin and these drugs was observed.
Effects on electrocardiogram: When healthy adults were administered a repeated oral dose of 40 mg or 160 mg of teneligliptin once daily for 4 days, a maximum mean (and upper limit of 90% confidence interval) of changes in QTcI (individually corrected QTc) interval corrected for placebo was 3.9 (7.6) msec in the 40 mg group 3 hours after the completion of administration and 9.3 (13.0) msec in the 160 mg group 1.5 hours after the completion of administration (see Precautions and Overdosage). [The usual approved dosage of this product is 20 mg of teneligliptin once daily, and the maximum dosage is 40 mg once daily (see Dosage & Administration).]
Toxicology: Preclinical safety data: In a 52-week repeated oral dose toxicity study in cynomolgus monkeys, skin lesions including exfoliation, scab and ulcer were observed on the tail, extremities and/or auricle at a dose of 75 mg/kg/day. AUC0-24hr when the lesions were observed reached approximately 45 times that in humans treated with 40 mg/day. The same toxicity findings have not been reported in other animal species (rats, mice and rabbits) and humans.
Teneligliptin was negative for genotoxicity in an in vitro bacterial reverse mutation test and in vivo micronucleus and unscheduled DNA synthesis tests in rats, although it was positive in an in vitro chromosomal aberration test due to secondary effects of cytotoxicity. Therefore, it is concluded that teneligliptin shows no genotoxicity.
Teneligliptin showed no carcinogenic potential in a 2-year carcinogenicity study in rats and a 26-week carcinogenicity study in transgenic mice.
In a fertility and early embryonic development study in rats, low body weight gain, decreased implantations and live embryos, and secondary changes in male reproductive organs due to low body weight gain were observed. In embryo-fetal development studies in rats and rabbits, skeletal variations and decreased ossifications were observed in fetuses, but no signs suggesting teratogenicity were noted. In a pre- and post-natal development study in rats, a slightly low body weight gain was observed in offspring. AUC0-24hr at the NOAELs in reproductive and developmental toxicity studies reached more than 10 times that in humans treated with 40 mg/day.