


 Sign Out
Sign Out


Hypersensitivity [see Contraindications and Precautions].
Hypocalcemia [see Contraindications and Precautions].
Osteonecrosis of the Jaw [see Osteonecrosis of the Jaw under Precautions].
Atypical Subtrochanteric and Diaphyseal Femoral Fractures [see Atypical Subtrochanteric and Diaphyseal Femoral Fractures under Precautions].
Clinical Trials Experience: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of EVENITY for the treatment of postmenopausal osteoporosis was evaluated in a multicenter, randomized, double-blind, placebo-controlled study (Study 1, NCT01575834) of 7180 postmenopausal women aged 55 to 90 years (mean age of 71 years). A total of 3581 and 3576 women received at least one dose of EVENITY and placebo, respectively, administered once every month during the 12-month double-blind study period. Women received at least 500 mg calcium and 600 international units of vitamin D supplementation daily and 77% received a loading dose of 50,000 to 60,000 international units of vitamin D within one week of randomization (if serum 25-hydroxyvitamin D concentrations were 40 ng/mL or less).
The safety of EVENITY for the treatment of postmenopausal osteoporosis in patients at high risk of fracture was evaluated in a multicenter, randomized, double-blind, alendronate-controlled study (Study 2, NCT01631214) of 4093 postmenopausal women aged 55 to 90 years (mean age of 74 years). A total of 2040 and 2014 women received at least one dose of EVENITY and alendronate, respectively, during the 12-month double-blind study period. Women received at least 500 mg calcium and 600 international units vitamin D supplementation daily and 74% received a loading dose of 50,000 to 60,000 international units of vitamin D within one week of randomization (if serum 25-hydroxyvitamin D concentrations were 40 ng/mL or less).
In Study 1, during the 12-month double-blind treatment period, the incidence of all-cause mortality was 0.7% (24/3576) in the placebo group and 0.8% (29/3581) in the EVENITY group. The incidence of nonfatal serious adverse events was 8.3% in the placebo group and 9.1% in the EVENITY group. The percentage of patients who withdrew from the study due to adverse events was 1.1% in the placebo group and 1.1% in the EVENITY group. The most common adverse reactions reported with EVENITY (greater than or equal to 5% and at a higher incidence than placebo) were arthralgia and headache. The most common adverse reaction leading to discontinuation of EVENITY was arthralgia (6 subjects [0.2%] in the placebo group and 5 subjects [0.1%] in the EVENITY group).
In Study 2, during the 12-month double-blind treatment period, the incidence of all-cause mortality was 1.1% (22/2014) in the alendronate group and 1.5% (30/2040) in the EVENITY group. The incidence of nonfatal serious adverse events was 13.3% in the alendronate group and 11.9% in the EVENITY group. The percentage of patients who withdrew from the study due to adverse events was 1.2% in the alendronate group and 1.2% in the EVENITY group. The most common adverse reactions reported with EVENITY (greater than or equal to 5%) were arthralgia and headache.
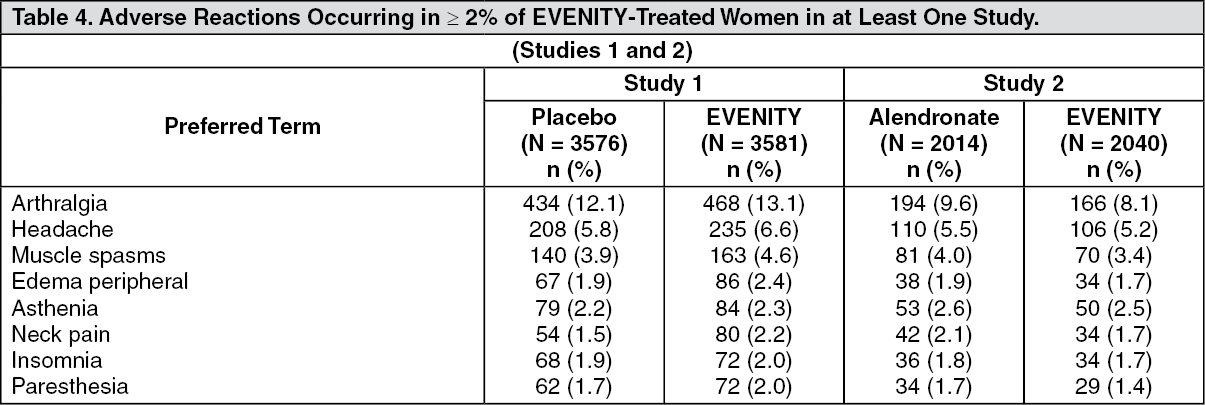
Table 4 outlines the most common adverse reactions occurring in greater than or equal to 2% of EVENITY-treated women in at least one study. (See Table 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe adverse reactions described as follows are from the 12-month treatment periods of Study 1 (placebo-controlled) and Study 2 (alendronate-controlled).
Major Adverse Cardiac Events (MACE): During the 12-month double-blind treatment period of the placebo-controlled trial (Study 1), myocardial infarction occurred in 9 women (0.3%) in the EVENITY group and 8 (0.2%) women in the placebo group; stroke occurred in 8 women (0.2%) in the EVENITY group and 10 (0.3%) women in the placebo group. These events occurred in patients with and without a history of myocardial infarction or stroke. Cardiovascular death occurred in 17 women (0.5%) in the EVENITY group and 15 (0.4%) women in the placebo group. The number of women with positively adjudicated MACE was 30 (0.8%) in the EVENITY group and 29 (0.8%) in the placebo group, yielding a hazard ratio of 1.03 (95% confidence interval [0.62, 1.72]) for EVENITY compared to placebo.
During the 12-month double-blind treatment period of the active-controlled trial (Study 2), myocardial infarction occurred in 16 women (0.8%) in the EVENITY group and 5 (0.2%) women in the alendronate group; stroke occurred in 13 women (0.6%) in the EVENITY group and 7 (0.3%) women in the alendronate group. These events occurred in patients with and without a history of myocardial infarction or stroke. Cardiovascular death occurred in 17 women (0.8%) in the EVENITY group and 12 (0.6%) women in the alendronate group. The number of women with positively adjudicated MACE was 41 (2.0%) in the EVENITY group and 22 (1.1%) in the alendronate group, yielding a hazard ratio of 1.87 (95% confidence interval [1.11, 3.14]) for EVENITY compared to alendronate [see Warnings and Major Adverse Cardiac Events (MACE) under Precautions].
Injection Site Reactions: Across both trials, injection site reactions occurred in 278 (4.9%) women in the EVENITY group and 157(2.8%) women in the control group. The most common injection site reactions were pain (94 [1.7%] women in the EVENITY group; 70 [1.3%] in the control group) and erythema (80 [1.4%] women in the EVENITY group and 14 [0.3%] women in the control group). Injection site reactions resulted in discontinuation of treatment in 7 (0.1%) EVENITY-treated patients and 3 (< 0.1%) patients in the control group.
Hypersensitivity Reactions: Across both trials, hypersensitivity reactions were reported in 364 (6.5%) women in the EVENITY group and 365 (6.5%) women in the control group. Reported reactions included angioedema (3 women [< 0.1%]in the EVENITY group vs. 3 [< 0.1%] women in the control group), erythema multiforme (1 woman [< 0.1%] in the EVENITY group vs. no woman in the control group), dermatitis (32 women [0.6%] in the EVENITY group vs. 42 women [0.8%] in the control group), rash (60 women [1.1%] in the EVENITY group vs. 53 women [0.9%] in the control group), and urticaria (23 women [0.4%] in the EVENITY group vs. 27 women [0.5%] in the control group). Although angioedema, dermatitis and urticaria were not reported at a higher incidence with EVENITY than control, there were cases of angioedema, dermatitis and urticaria that were determined to be related to EVENITY use [see Contraindications and Hypersensitivity Reactions under Precautions].
Hypocalcemia: Across both trials, adverse events of hypocalcemia occurred in 2 EVENITY-treated women and in 1 woman in the control group. Decreases in albumin-adjusted serum calcium to below the lower limit of the reference range (8.3 mg/dL) were reported in 14 (0.2%) women in the EVENITY group and 10 (0.2%) women in the control group. No patient receiving EVENITY developed serum calcium less than 7.5 mg/dL. The nadir in albumin-adjusted serum calcium occurred by month 1 after EVENITY dosing in patients with normal renal function [see Contraindications and Hypocalcemia under Precautions].
Osteonecrosis of the Jaw: Across both trials, osteonecrosis of the jaw occurred in one patient during treatment with EVENITY. [see Osteonecrosis of the Jaw under Precautions].
Atypical Subtrochanteric and Diaphyseal Fractures: Across both trials, atypical femoral fracture occurred in one patient during treatment with EVENITY [see Atypical Subtrochanteric and Diaphyseal Femoral Fractures under Precautions].
Immunogenicity: As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described as follows with the incidence of antibodies in other studies or to other romosozumab products may be misleading.
The immunogenicity of EVENITY was evaluated using an immunoassay for the detection of anti-romosozumab antibodies. An in vitro biological assay was performed to detect neutralizing antibodies for those subjects whose sera tested positive for anti-romosozumab antibodies.
Among 5914 postmenopausal women treated with EVENITY 210 mg monthly, 18.1% of subjects developed antibodies to romosozumab. Of the subjects who developed antibodies to romosozumab, 4.7% had antibodies that were classified as neutralizing. Development of antibodies to romosozumab was associated with lower serum romosozumab concentrations [see Pharmacology: Pharmacokinetics under Actions].
Antibodies to romosozumab were generally not associated with changes in the efficacy or safety of EVENITY.
View ADR Monitoring Form