


 Sign Out
Sign Out



PHARMACOLOGY: Pharmacodynamics: Mechanism of action: Dolutegravir inhibits HIV integrase by binding to the integrase active site and blocking the strand transfer step of retroviral Deoxyribonucleic acid (DNA) integration which is essential for the HIV replication cycle. Strand transfer biochemical assays using purified HIV-1 integrase and pre-processed substrate DNA resulted in IC50 values of 2.7 nM and 12.6 nM. In vitro, dolutegravir dissociates slowly from the active site of the wild type integrase-DNA complex (t½ 71 hours).
Lamivudine is a NRTI, and is a potent, selective inhibitor of HIV-1 and HIV-2. Lamivudine is metabolised sequentially by intracellular kinases to the respective triphosphate (TP) which is the active moiety with an extended intracellular half-life supporting once daily dosing (see Pharmacokinetics: Elimination as follows). Lamivudine-TP is a substrate for and competitive inhibitor of HIV reverse transcriptase (RT). However, its main antiviral activity is through incorporation of the monophosphate form into the viral DNA chain, resulting in chain termination. Lamivudine-TP shows significantly less affinity for host cell DNA polymerases.
Pharmacodynamic Effects: In a randomized, dose-ranging trial, HIV 1-infected subjects treated with dolutegravir monotherapy (ING111521) demonstrated rapid and dose-dependent antiviral activity, with mean declines from baseline to day 11 in HIV-1 RNA of 1.5, 2.0, and 2.5 log10 for dolutegravir 2 mg, 10 mg, and 50 mg once daily, respectively. This antiviral response was maintained for 3 to 4 days after the last dose in the 50 mg group.
Antiviral Activity in cell culture: Dolutegravir exhibited antiviral activity against laboratory strains of wild-type HIV-1 with mean concentration of drug necessary to effect viral replication by 50 percent (EC50) values of 0.5 nM (0.21 ng per mL) to 2.1 nM (0.85 ng per mL) in peripheral blood mononuclear cells (PBMCs) and MT-4 cells.
In a viral integrase susceptibility assay using the integrase coding region from 13 clinically diverse clade B isolates, dolutegravir demonstrated antiviral potency similar to laboratory strains, with a mean EC50 of 0.52 nM. When tested in PBMC assays against a panel consisting of 24 HIV-1 clinical isolates [group M (clade A, B, C, D, E, F and G) and group O] and 3 HIV-2 clinical isolates, the geometric mean EC50 was 0.20 nM and EC50 values ranged from 0.02 to 2.14 nM for HIV-1, while the geometric mean EC50 was 0.18 nM and EC50 values ranged from 0.09 to 0.61 nM for HIV-2 isolates.
The antiviral activity of lamivudine against HIV-1 was assessed in a number of cell lines including monocytes and PBMCs using standard susceptibility assays. EC50 values were in the range of 0.003 to 0.17 μM. The EC50 values of lamivudine against different HIV-1 clades (A-G) ranged from 0.001 to 0.120 μM, and against HIV-2 isolates from 0.002 to 0.120 μM in PBMCs.
Antiviral Activity in combination with other antiviral agents: No drugs with inherent anti-HIV activity were antagonistic with dolutegravir (in vitro assessments were conducted in checkerboard format in combination with stavudine, abacavir, efavirenz, nevirapine, lopinavir, amprenavir, enfuvirtide, maraviroc, adefovir and raltegravir). In addition, antivirals without inherent anti-HIV activity (ribavirin) had no apparent effect on dolutegravir activity.
No antagonistic effects in vitro were seen with lamivudine and other antiretrovirals (tested agents: abacavir, didanosine, nevirapine, zalcitabine, and zidovudine).
Effect of Human Serum and Serum Proteins: In vitro studies suggested a 75-fold shift in EC50 of dolutegravir in the presence of 100% human serum (by method of extrapolation), and the protein adjusted EC90 (PA-EC90) in PBMCs was estimated to be 64 ng/mL. Dolutegravir trough concentration for a single 50 mg dose in integrase inhibitor naïve subjects was 1.20 µg/mL, 19 times higher than the estimated PA-EC90. Lamivudine exhibits linear pharmacokinetics over the therapeutic dose range and displays low plasma protein binding (less than 36%).
Resistance in vitro and in vivo (dolutegravir): Viruses highly resistant to dolutegravir were not observed during the 112 day passage of strain IIIB, with a 4.1-fold maximum fold change (FC) observed for the passaged resistant virus populations with substitutions at the conserved IN positions S153Y and S153F. Passage of the wild type HIV-1 strain NL432 in the presence of dolutegravir selected for E92Q (passage population virus FC=3.1) and G193E (passage population virus FC=3.2) substitutions on Day 56. Additional passage of wild type clade B, C, and A/G viruses in the presence of dolutegravir selected for G118R (site-directed mutant FC=10), S153T, and R263K (site-directed mutant FC=1.5).
Treatment-naïve HIV-1 infected subjects receiving dolutegravir: No INI-resistant mutations or treatment emergent resistance to the NRTI backbone therapy were isolated with dolutegravir 50 mg once daily in treatment-naive studies.
Resistance in vitro and in vivo (lamivudine): HIV-1 resistance to lamivudine involves the development of a M184I or M184V amino acid change close to the active site of the viral RT. This variant arises both during in vitro selection and in HIV-1 infected patients treated with lamivudine-containing antiretroviral therapy. M184V mutants display greatly reduced susceptibility to lamivudine and show diminished viral replicative capacity in vitro.
Resistance in vivo (dolutegravir plus lamivudine): No subjects that met the protocol-defined confirmed virologic withdrawal (CVW) criteria across the pooled GEMINI-1 and GEMINI-2 studies through Week 144 or in the TANGO study through Week 96 had emergent INSTI or NRTI resistance substitutions. In patients with prior failed therapies, but naïve to the integrase class (SAILING study), integrase inhibitor substitutions were observed in 4/354 patients (follow-up 48 weeks) treated with dolutegravir, which was given in combination with an investigator selected background regimen. Of these four, two subjects had a unique R263K integrase substitution, with a maximum fold change of 1.93, one subject had a polymorphic V151V/I integrase substitution, with fold change of 0.92, and one subject had pre-existing integrase mutations and is assumed to have been integrase experienced or infected with integrase resistant virus by transmission. In patients naïve to the integrase class and failing first line NNRTI + 2 NRTI treatment (DAWNING study) through Week 48, 2/314 subjects treated with dolutegravir had integrase inhibitor G118R pathway substitutions conferring dolutegravir fold changes of 15 and 30, and respective viral replication capacity decreases of 6.6 fold and 18 fold compared with baseline. The G118R and R263K mutations were also selected in vitro (see previous text).
Cross-resistance: Site-directed INSTI mutant virus: Dolutegravir activity was determined against a panel of 60 INSTI-resistant site-directed mutant HIV-1 viruses (28 with single substitutions and 32 with 2 or more substitutions). The single INSTI-resistance substitutions T66K, I151L, and S153Y conferred a greater than 2-fold decrease in dolutegravir susceptibility (range: 2.3-fold to 3.6-fold from reference). Combinations of multiple substitutions T66K/L74M, E92Q/N155H, G140C/Q148R, G140S/Q148H, R or K, Q148R/N155H, T97A/G140S/Q148, and substitutions at E138/G140/Q148 showed a greater than 2-fold decrease in dolutegravir susceptibility (range: 2.5-fold to 21-fold from reference).
Recombinant clinical isolates: Dolutegravir activity was measured for 705 raltegravir resistant recombinant isolates from clinical practice; 93.9% (662/705) of the isolates had a dolutegravir FC ≤10 and 1.8% had a dolutegravir FC >25. Mutants with Y143 and N155 pathway had mean FCs of 1.2 and 1.5, respectively, while Q148 + 1 mutant and Q148 + ≥2 mutants mean FCs were 4.8 and 6.0, respectively.
Cross-resistance conferred by the M184V reverse transcriptase: Cross-resistance is limited within the nucleoside inhibitor class of antiretroviral agents. Zidovudine and stavudine maintain their antiretroviral activities against lamivudine-resistant HIV-1. Abacavir and tenofovir maintain antiretroviral activity against lamivudine-resistant HIV-1 harbouring only the M184V mutation.
Effects on Electrocardiogram: In a randomised, placebo-controlled, cross-over trial, 42 healthy subjects received single dose oral administrations of placebo, dolutegravir 250 mg suspension (exposures approximately 3-fold of the 50 mg once-daily dose at steady state), and moxifloxacin (400 mg, active control) in random sequence. Dolutegravir did not prolong the QTc interval for 24 hours post dose. After baseline and placebo adjustment, the maximum mean QTc change based on Fridericia correction method (QTcF) was 1.99 msec (1-sided 95% upper CI: 4.53 msec).
Similar studies were not conducted with lamivudine.
Effects on Renal Function: The effect of dolutegravir on serum creatinine clearance (CrCl), glomerular filtration rate (GFR) using iohexol as the probe and effective renal plasma flow (ERPF) using paraaminohippurate (PAH) as the probe was evaluated in an open-label, randomised, 3 arm, parallel, placebo-controlled study in 37 healthy subjects, who were administered dolutegravir 50 mg once daily (n=12), 50 mg twice daily (n=13) or placebo once daily (n=12) for 14 days. A modest decrease in CrCl was observed with dolutegravir within the first week of treatment, consistent with that seen in clinical studies. Dolutegravir at both doses had no significant effect on GFR or ERPF. These data support in vitro studies which suggest that the small increases in creatinine observed in clinical studies are due to the nonpathologic inhibition of the organic cation transporter 2 (OCT2) in the proximal renal tubules, which mediates the tubular secretion of creatinine.
In the pooled analysis of GEMINI-1 and GEMINI-2 studies in treatment-naïve adult patients at the week 144 analysis, dolutegravir plus lamivudine was associated with lower impact on renal safety parameters compared to dolutegravir plus tenofovir/emtricitabine FDC. The dolutegravir plus lamivudine group had a significantly greater increase in the estimated GFR using cystatin C adjusted CKD-EPI equation, compared with the dolutegravir plus tenofovir/emtricitabine FDC group (adjusted mean change from baseline of 12.2 and 10.6 mL/min/1.73 m2, respectively; p=0.008). Change from baseline analysis showed that urine albumin/creatinine and protein/creatinine ratios were lower in the dolutegravir plus lamivudine group compared with the dolutegravir plus tenofovir/emtricitabine FDC group; the difference was statistically significant for the protein/creatinine ratio (urine albumin/creatinine week 144/baseline ratio of 1.046 and 1.104, respectively; p = 0.261 and protein/creatinine week 144/baseline ratio of 0.994 and 1.193, respectively; p <0.001). Withdrawals from study for renal function-related adverse events or for meeting predefined renal toxicity criteria (eGFR <50 ml/min/1.73 m2) were more frequently observed in subjects in the dolutegravir plus tenofovir/emtricitabine FDC group compared with the dolutegravir plus lamivudine group.
Clinical Studies: Antiretroviral naïve subjects: The efficacy of DOVATO is supported by data from 2 identical 148-week, Phase III, randomised, double-blind, multicenter, parallel-group, non-inferiority controlled trials (GEMINI-1 [204861] and GEMINI-2 [205543]). A total of 1,433 HIV-1 infected antiretroviral treatment-naïve adult subjects received treatment in the trials. Subjects were enrolled with a screening plasma HIV-1 RNA of 1,000 c/mL to ≤500,000 c/mL. Subjects were randomised to a two-drug regimen of dolutegravir plus lamivudine administered once daily or dolutegravir plus tenofovir/emtricitabine FDC administered once daily. The primary efficacy endpoint for each GEMINI trial was the proportion of subjects with plasma HIV-1 RNA <50 copies/mL at Week 48 (Snapshot algorithm for the ITT-E population).
At baseline, in the pooled analysis, the median age of subjects was 33 years, 15% were female, 69% were white, 9% were CDC Stage 3 (AIDS), 20% had HIV-1 RNA >100,000 copies/mL, and 8% had CD4+ cell count less than 200 cells per mm3; these characteristics were similar between studies and treatment arms.
In the primary week 48 analysis, dolutegravir plus lamivudine was non-inferior to dolutegravir plus tenofovir/emtricitabine FDC in GEMINI-1 and GEMINI-2 studies. This was supported by the pooled analysis, see Table 1.
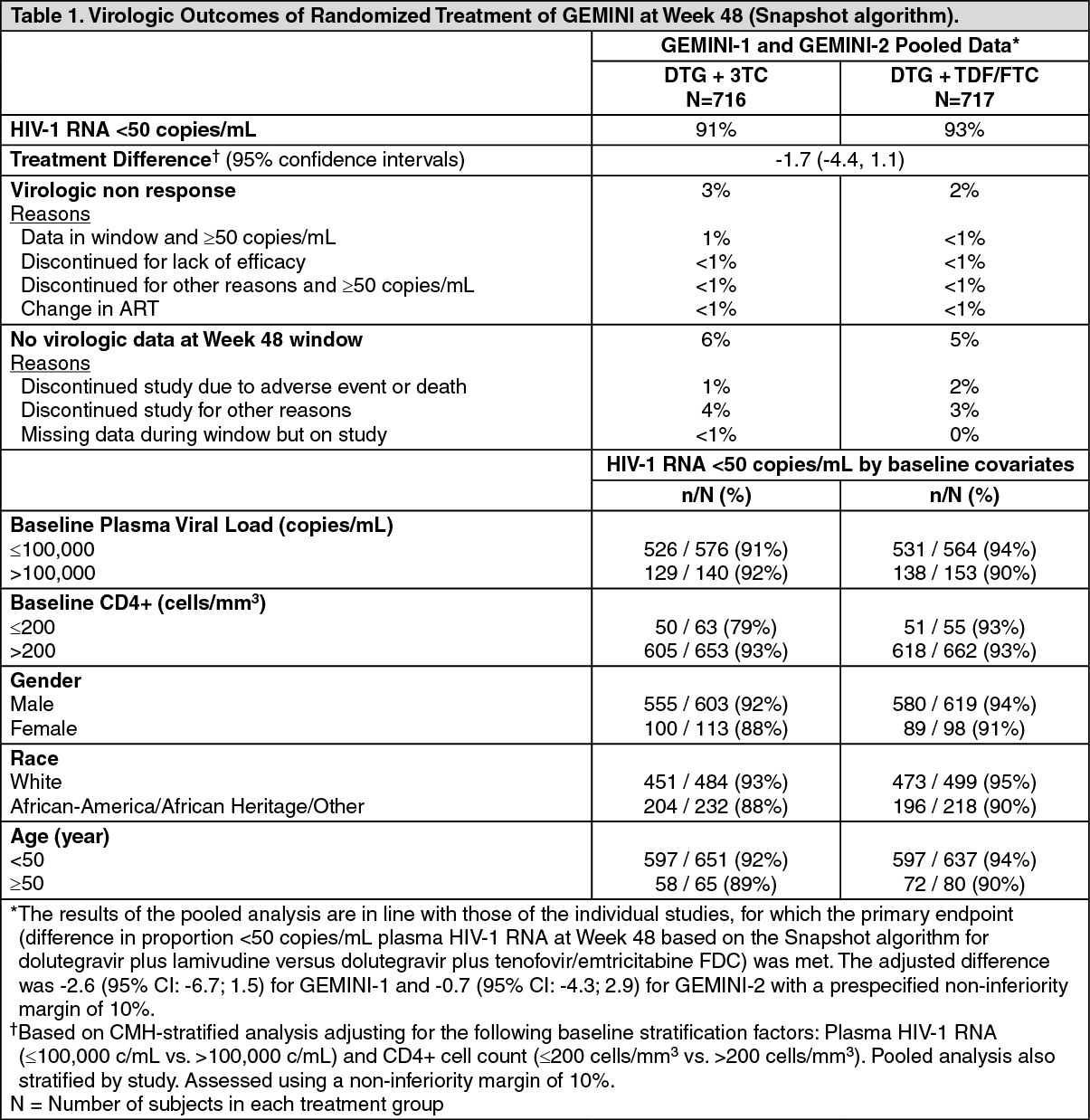 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAt 96 weeks in the GEMINI-1 and GEMINI-2 studies, the dolutegravir plus lamivudine group (86% with plasma HIV-1 RNA < 50 copies/mL [pooled data]) remained non-inferior to the dolutegravir plus tenofovir/emtricitabine FDC group (90% with plasma HIV-1 RNA < 50 copies/mL [pooled data]). The adjusted difference in proportions and 95% CI was -3.4% (-6.7, 0.0). The results of the pooled analysis were in line with those of the individual studies, for which the secondary endpoint (difference in proportion <50 copies/mL plasma HIV-1 RNA at Week 96 based on the Snapshot algorithm for dolutegravir plus lamivudine versus dolutegravir plus tenofovir/emtricitabine FDC) was met. The adjusted differences of -4.9 (95% CI: -9.8; 0.0) for GEMINI-1 and -1.8 (95% CI: -6.4; 2.7) for GEMINI-2 were within the prespecified non-inferiority margin of -10%.
The mean increase in CD4+ T-cell counts was 269 cells/mm3 in the DTG+3TC arm and 259 cells/mm3 in the DTG+FTC/TDF arm, at week 96.
At 144 weeks in the GEMINI-1 and GEMINI-2 studies, the dolutegravir plus lamivudine group (82% with plasma HIV-1 RNA < 50 copies/mL [pooled data]) remained non-inferior to dolutegravir plus tenofovir/emtricitabine FDC group (84% with plasma HIV-1 RNA < 50 copies/mL [pooled data]). The results of the pooled analysis were in line with those of the individual studies, for which the secondary endpoint (difference in proportion < 50 copies/mL plasma HIV-1 RNA at Week 144 based on the Snapshot algorithm for dolutegravir plus lamivudine versus dolutegravir plus tenofovir/emtricitabine FDC) was met. The adjusted difference in proportions and 95% CI for the pooled data was -1.8% (-5.8, 2.1). The adjusted differences of -3.6 (95% CI: -9.4, 2.1) for GEMINI-1 and 0.0 (95% CI: -5.3, 5.3) for GEMINI-2 were within the prespecified non-inferiority margin of -10%.
The mean increase in CD4+ T-cell counts was 302 cells/mm3 in the DTG+3TC arm and 300 cells/mm3 in the DTG+FTC/TDF arm, at Week 144.
Virologically suppressed subjects: The efficacy of DOVATO in HIV-infected, antiretroviral therapy experienced, virologically suppressed subjects is supported by data from a 200-week, Phase III, randomised, open-label, multicenter, parallel-group, non-inferiority controlled trial (TANGO [204862]). A total of 741 adult HIV-1 infected subjects who were on a stable suppressive tenofovir alafenamide based regimen (TBR) received treatment in the studies. Subjects were randomised in a 1:1 ratio to receive DOVATO once daily or continue with TBR for up to 200 weeks. Randomisation was stratified by baseline third agent class (protease inhibitor [PI], integrase inhibitor [INSTI], or non-nucleoside reverse transcriptase inhibitor [NNRTI]). The primary efficacy endpoint was the proportion of subjects with plasma HIV-1 RNA ≥50 c/mL (virologic non-response) as per the FDA Snapshot category at Week 48 (Snapshot algorithm adjusting for randomization stratification factor: Baseline Third Agent Class [INSTI, PI, NNRTI]).
At baseline the median age of subjects was 39 years, 8% were female and 21% nonwhite, 5% were CDC Class C (AIDS) and 98% subjects had Baseline CD4+ cell count ≥200 cells/mm3; these characteristics were similar between treatment arms. Subjects had been on ART for a median of 2.8 years and 2.9 years prior to Day 1 for the DOVATO and TBR arms, respectively. Most subjects were on INSTI-based TBR, 78% and 80% in the DOVATO and TBR arms, respectively.
In the primary 48 week analysis, DOVATO was non-inferior to TBR, with <1% of subjects in both arms experiencing virologic failure (HIV-1 RNA ≥50 c/mL) based on the Snapshot algorithm (Table 2). (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAt 96 weeks in the TANGO study, the proportion of subjects with HIV-1 RNA ≥50 c/mL (Snapshot) was 0.3% and 1.1% in the DOVATO and TBR groups, respectively. Based on a non-inferiority margin of 4%, DOVATO remained non-inferior to TBR, as the upper bound of the 95% CI for the adjusted treatment difference (-2.0%, 0.4%) was less than 4% for the ITT E Population.
The median change from baseline in CD4+ T-cell counts at Week 96 was 61 cells/mm3 in the DOVATO arm and 45 cells/mm3 in the TBR arm.
Antiretroviral Pregnancy Registry: The APR has received reports of over 600 exposures to dolutegravir during pregnancy resulting in live births, as of July 2019. These consist of over 370 exposures during the first trimester, over 230 exposures during the second/third trimester and included 12 and 9 birth defects, respectively. The prevalence (95% CI) of defects among live births exposed to dolutegravir in the first trimester was 3.2% (1.7%, 5.5%) and in the second/third trimester, 3.8% (1.7%, 7.0%).
The APR has received reports of over 12,500 exposures to lamivudine during pregnancy resulting in live birth, as of July 2019. These consist of over 5,200 exposures during the first trimester, over 7,400 exposures during the second/third trimester and included 161 and 216 birth defects, respectively. The prevalence (95% CI) of defects among live births exposed to lamivudine in the first trimester was 3.1% (2.6, 3.6%) and in the second/third trimester, 2.9% (2.5, 3.3%).
The available data from the APR shows no significant increase in risk of major birth defects for dolutegravir or lamivudine compared to the background rates in the two population based surveillance systems (Metropolitan Atlanta Congenital Defects Program with defects of 2.72 per 100 live births and the Texas Birth Defects Registry with 4.17 per 100 live births).
Children: There are no clinical study data with DOVATO in the paediatric population.
In a Phase I/II 48 week multicentre, open-label study (P1093/ING112578), the pharmacokinetic parameters, safety, tolerability and efficacy of dolutegravir was evaluated in combination regimens in HIV-1 infected infants, children and adolescents.
At 24 weeks, 16 of 23 (69%) adolescents (12 to less than 18 years of age) treated with dolutegravir once daily (35 mg n=4, 50 mg n=19) plus OBR achieved viral load less than 50 copies/mL.
Pharmacokinetics: When administered in fasted state, bioequivalence was achieved for dolutegravir, when comparing the DOVATO tablet to dolutegravir 50 mg co-administered with lamivudine 300 mg, for AUC and Cmax.
When administered in fasted state, bioequivalence was achieved for lamivudine AUC, when comparing the DOVATO tablet to lamivudine 300 mg co-administered with dolutegravir 50 mg. Lamivudine Cmax for the DOVATO tablet was 32% higher than lamivudine 300 mg co-administered with dolutegravir 50 mg. Following multiple oral doses of DOVATO in HIV-infected, treatment experienced subjects in the Phase III TANGO study, the steady state dolutegravir and lamivudine AUC and Cmax were similar to historical exposures.
Absorption: Dolutegravir and lamivudine are rapidly absorbed following oral administration. The absolute bioavailability of dolutegravir has not been established. The absolute bioavailability of oral lamivudine in adults is 80 to 85%. For DOVATO, the median time to maximal plasma concentrations (tmax) is 2.5 hours for dolutegravir and 1.0 hour for lamivudine, when dosed under fasted conditions.
Following multiple oral doses of dolutegravir 50 mg once daily, the geometric mean steady state pharmacokinetic parameter estimates are 53.6 µg·h/mL for AUC24, 3.67 µg/mL for Cmax, and 1.11 µg/mL for C24. Following multiple-dose oral administration of lamivudine 300 mg once daily for seven days the mean steady-state Cmax is 2.04 µg/mL and the mean AUC24 is 8.87 µg·h/mL.
Effect of Food: Administration of DOVATO with a high fat meal increased dolutegravir AUC and Cmax by 33% and 21%, respectively, and decreased the lamivudine Cmax by 30% compared to fasted conditions. The lamivudine AUC was not affected by a high fat meal. These changes are not clinically significant. DOVATO may be administered with or without food.
Distribution: The apparent volume of distribution of dolutegravir (following oral administration of suspension formulation, Vd/F) is estimated at 12.5 L. Intravenous studies with lamivudine showed that the mean apparent volume of distribution is 1.3 L/kg.
Dolutegravir is highly bound (approximately 99.3%) to human plasma proteins based on in vitro data. Binding of dolutegravir to plasma proteins was independent of concentration. Total blood and plasma drug-related radioactivity concentration ratios averaged between 0.441 to 0.535, indicating minimal association of radioactivity with blood cellular components. Free fraction of dolutegravir in plasma is estimated at approximately 0.2 to 1.1% in healthy subjects, approximately 0.4 to 0.5% in subjects with moderate hepatic impairment, and 0.8 to 1.0% in subjects with severe renal impairment and 0.5% in HIV-1 infected patients. Lamivudine exhibits linear pharmacokinetics over the therapeutic dose range and displays low plasma protein binding (less than 36%).
Dolutegravir and lamivudine are present in cerebrospinal fluid (CSF). In 12 treatment-naïve subjects receiving a regimen of dolutegravir plus abacavir/lamivudine for 16 weeks, dolutegravir concentration in CSF averaged 15.4 ng/mL at Week 2 and 12.6 ng/mL at Week 16, ranging from 3.7 to 23.2 ng/mL (comparable to unbound plasma concentration). CSF:plasma concentration ratio of dolutegravir ranged from 0.11 to 2.04%. Dolutegravir concentrations in CSF exceeded the IC50, supporting the median reduction from baseline in CSF HIV-1 RNA of 2.2 log after 2 weeks and 3.4 log after 16 weeks of therapy (see Pharmacodynamics as previously mentioned). The mean ratio of CSF/serum lamivudine concentrations 2 to 4 h after oral administration was approximately 12%. The true extent of CNS penetration of lamivudine and its relationship with any clinical efficacy is unknown.
Dolutegravir is present in the female and male genital tract. AUC in cervicovaginal fluid, cervical tissue, and vaginal tissue were 6 to 10% of that in corresponding plasma at steady-state. AUC was 7% in semen and 17% in rectal tissue, of those in corresponding plasma at steady-state.
Metabolism: Dolutegravir is primarily metabolized via UGT1A1 with a minor CYP3A component (9.7% of total dose administered in a human mass balance study). Dolutegravir is the predominant circulating compound in plasma; renal elimination of unchanged drug is low (<1% of the dose). Fifty-three percent of total oral dose is excreted unchanged in the faeces. It is unknown if all or part of this is due to unabsorbed drug or biliary excretion of the glucuronidate conjugate, which can be further degraded to form the parent compound in the gut lumen. Thirty-one percent of the total oral dose is excreted in the urine, represented by ether glucuronide of dolutegravir (18.9% of total dose), N-dealkylation metabolite (3.6% of total dose), and a metabolite formed by oxidation at the benzylic carbon (3.0% of total dose).
Metabolism of lamivudine is a minor route of elimination. Lamivudine is predominately cleared unchanged by renal excretion. The likelihood of metabolic interactions with lamivudine is low due to the small extent of hepatic metabolism (less than 10%).
Elimination: Dolutegravir has a terminal half-life of ~14 hours and an apparent clearance (CL/F) of 0.56 L/hr.
The observed half-life of elimination for lamivudine is 18 to 19 hours. For patients receiving lamivudine 300 mg once daily, the terminal intracellular half-life of lamivudine-TP was 16 to 19 hours. The mean systemic clearance of lamivudine is approximately 0.32 L/h/kg, predominantly by renal clearance (greater than 70%) via the organic cationic transport system.
Special patient populations: Children: DOVATO has not been studied in the paediatric population.
In a paediatric study including 23 antiretroviral treatment-experienced HIV-1 infected adolescents aged 12 to 18 years of age, the pharmacokinetics of dolutegravir was evaluated in 10 adolescents and showed that dolutegravir 50 mg once daily dosage resulted in dolutegravir exposure in paediatric subjects comparable to that observed in adults who received dolutegravir 50 mg once daily (Table 3). (See Table 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageLimited data are available in adolescents receiving a daily dose of 300 mg of lamivudine. Pharmacokinetic parameters are comparable to those reported in adults.
Elderly: Population pharmacokinetic analysis of dolutegravir using data in HIV-1 infected adults showed that there was no clinically relevant effect of age on dolutegravir exposure.
Pharmacokinetic data for dolutegravir and lamivudine in subjects of >65 years old are limited.
Renal impairment: Pharmacokinetic data have been obtained for dolutegravir and lamivudine alone. DOVATO should not be used in patients with creatinine clearance of less than 30 mL/min because, whilst no dosage adjustment of dolutegravir is necessary in patients with renal impairment, dose reduction is required for the lamivudine component.
Studies with lamivudine show that plasma concentrations (AUC) are increased in patients with renal dysfunction due to decreased clearance.
Renal clearance of unchanged drug is a minor pathway of elimination for dolutegravir. A study of the pharmacokinetics of dolutegravir was performed in subjects with severe renal impairment (CLcr <30 mL/min). No clinically important pharmacokinetic differences between subjects with severe renal impairment (CLcr <30 mL/min) and matching healthy subjects were observed.
Hepatic impairment: Pharmacokinetic data has been obtained for dolutegravir and lamivudine individually.
Data obtained for lamivudine in patients with moderate to severe hepatic impairment and for dolutegravir in patients with moderate hepatic impairment show that the pharmacokinetics are not significantly affected by hepatic dysfunction. Dolutegravir is primarily metabolized and eliminated by the liver. In a study comparing 8 subjects with moderate hepatic impairment (Child-Pugh category B) to 8 matched healthy adult controls, the single 50 mg dose exposure of dolutegravir was similar between the two groups. The effect of severe hepatic impairment on the pharmacokinetics of dolutegravir has not been studied.
Polymorphisms in Drug Metabolising Enzymes: There is no evidence that common polymorphisms in drug metabolising enzymes alter dolutegravir pharmacokinetics to a clinically meaningful extent. In a meta-analysis using pharmacogenomics samples collected in clinical studies in healthy subjects, subjects with UGT1A1 (n=7) genotypes conferring poor dolutegravir metabolism had a 32% lower clearance of dolutegravir and 46% higher AUC compared with subjects with genotypes associated with normal metabolism via UGT1A1 (n=41). Polymorphisms in CYP3A4, CYP3A5, and NR1I2 were not associated with differences in the pharmacokinetics of dolutegravir.
Gender: Population pharmacokinetic analyses revealed no clinically relevant effect of gender on the exposure of dolutegravir.
No clinically relevant differences in the pharmacokinetics of lamivudine have been observed between men and women.
Race: Population pharmacokinetic analyses using pooled pharmacokinetic data from Phase IIb and Phase III adult trials for dolutegravir revealed no clinically relevant effect of race on the exposure of dolutegravir. The pharmacokinetics of dolutegravir following single dose oral administration to Japanese subjects appear similar to observed parameters in Western (US) subjects.
There is no evidence that a dose adjustment of dolutegravir or lamivudine would be required based on the effects of race on PK parameters.
Co-infection with Hepatitis B or C: Population pharmacokinetic analysis indicated that hepatitis C virus co-infection had no clinically relevant effect on the exposure to dolutegravir. There are limited pharmacokinetic data on subjects with hepatitis B co-infection (see Precautions).
Pregnancy: The pharmacokinetics of lamivudine during pregnancy are similar to that of non-pregnant adults. In humans, consistent with passive transmission of lamivudine across the placenta, lamivudine concentrations in infant serum at birth were similar to those in maternal and cord serum at delivery.
There are no pharmacokinetic data on the use of dolutegravir in pregnancy.
Toxicology: Pre-clinical Safety Data: Carcinogenesis/mutagenesis: Dolutegravir was not mutagenic or clastogenic using in vitro tests in bacteria and cultured mammalian cells, and an in vivo rodent micronucleus assay. Dolutegravir was not carcinogenic in long term studies in the mouse and rat.
Lamivudine was not mutagenic in bacterial tests, but like many nucleoside analogues it shows activity in the in vitro mammalian tests such as the mouse lymphoma assay. This is consistent with the known activity of other nucleoside analogues. The results from two in vivo rat micronucleus tests with lamivudine were negative.
Lamivudine did not show any genotoxic activity in additional in vivo studies in rats (metaphase analysis of bone marrow and unscheduled DNA synthesis). The results of long-term carcinogenicity studies in mice and rats did not show any carcinogenic potential at exposures approximately 12 to 72 times higher than clinical plasma levels.
Reproductive Toxicology: Fertility: Fertility studies in the rat have shown that dolutegravir and lamivudine had no effect on male or female fertility.
Dolutegravir did not affect male or female fertility in rats at doses up to 1,000 mg/kg/day, the highest dose tested (33 times the 50 mg human clinical exposure, based on AUC).
Pregnancy: Dolutegravir and lamivudine were shown to cross the placenta in animal reproductive toxicity studies.
Oral administration of dolutegravir to pregnant rats at doses up to 1,000 mg/kg daily from days 6 to 17 of gestation did not elicit maternal toxicity, developmental toxicity or teratogenicity (37.9 times the 50 mg human clinical exposure, based on AUC).
Oral administration of dolutegravir to pregnant rabbits at doses up to 1,000 mg/kg daily from days 6 to 18 of gestation did not elicit developmental toxicity or teratogenicity (0.56 times the 50 mg human clinical exposure, based on AUC). In rabbits, maternal toxicity (decreased food consumption, scant/no faeces/urine, suppressed body weight gain) was observed at 1,000 mg/kg (0.56 times the 50 mg human clinical exposure, based on AUC).
Lamivudine was not teratogenic in animal studies, but there were indications of an increase in early embryonic deaths in rabbits at exposure levels comparable to those achieved in man. However, there was no evidence of embryonic loss in rats at exposure levels of approximately 32 times the clinical exposure (based on Cmax).
Animal toxicology and/or pharmacology: The effect of prolonged daily treatment with high doses of dolutegravir has been evaluated in repeat oral dose toxicity studies in rats (up to 26 weeks) and in monkeys (up to 38 weeks). The primary effect of dolutegravir was gastrointestinal intolerance or irritation in rats and monkeys at doses that produce systemic exposures approximately 30 and 1.2 times the 50 mg human clinical exposure based on AUC, respectively. Because gastrointestinal intolerance is considered to be due to local drug administration, mg/kg or mg/m2 metrics are appropriate determinates of safety cover for this toxicity. GI intolerance in monkeys occurred at 30 times the human mg/kg equivalent dose (based on 50 kg human), and 11 times the human mg/m2 equivalent dose for a total daily clinical dose of 50 mg.