Pharmacology: Pharmacodynamics: Mechanism of Action: Tofacitinib is a potent, selective inhibitor of the JAK family of kinases with a high degree of selectivity against other kinases in the human genome. In kinase assays, tofacitinib inhibits JAK1, JAK2, JAK3, and to a lesser extent TyK2. In cellular settings where JAK kinases signal in pairs, tofacitinib preferentially inhibits signaling by heterodimeric receptors associated with JAK3 and/or JAK1 with functional selectivity over receptors that signal via pairs of JAK2. Inhibition of JAK1 and JAK3 by tofacitinib blocks signaling through the common gamma chain-containing receptors for several cytokines, including IL-2, -4, -7, -9, -15, and -21. These cytokines are integral to lymphocyte activation, proliferation, and function and inhibition of their signaling may thus result in modulation of multiple aspects of the immune response. In addition, inhibition of JAK1 will result in attenuation of signaling by additional pro-inflammatory cytokines, such as IL-6 and Type I interferons. At higher exposures, inhibition of erythropoietin signaling could occur via inhibition of JAK2 signaling.
Pharmacodynamic Effect: In patients with rheumatoid arthritis, treatment up to 6 months with tofacitinib was associated with dose-dependent reductions of circulating CD16/56+ natural killer (NK) cells, with estimated maximum reductions occurring at approximately 8-10 weeks after initiation of therapy. These changes generally resolved within 2-6 weeks after discontinuation of treatment. Treatment with tofacitinib was associated with dose-dependent increases in B cell counts. Changes in circulating T-lymphocyte counts and T-lymphocyte subsets (CD3+, CD4+ and CD8+) were small and inconsistent.
Following long-term treatment (median duration of tofacitinib treatment of approximately 5 years), CD4+ and CD8+ counts showed median reductions of 28% and 27%, respectively, from baseline. In contrast to the observed decrease after short-term dosing, CD16/56+ natural killer cell counts showed a median increase of 73% from baseline. CD19+ B cell counts showed no further increases after long-term tofacitinib treatment. These changes returned toward baseline after temporary discontinuation of treatment. There was no evidence of an increased risk of serious or opportunistic infections or herpes zoster at low values of CD4+, CD8+ or NK cell counts or high B cell counts.
Changes in total serum IgG, IgM, and IgA levels over 6-month tofacitinib dosing in patients with rheumatoid arthritis were small, not dose-dependent and similar to those seen on placebo.
After treatment with tofacitinib in patients with rheumatoid arthritis, rapid decreases in serum C-reactive protein (CRP) were observed and maintained throughout dosing. Changes in CRP observed with tofacitinib treatment do not reverse fully within 2 weeks after discontinuation, indicating a longer duration of pharmacodynamic activity compared to the half-life.
Similar changes in T cells, B cells and serum CRP have been observed in patients with active psoriatic arthritis, although reversibility was not assessed. Total serum immunoglobulins were not assessed in patients with active psoriatic arthritis.
Clinical Safety: In one large randomized open-label PASS in RA patients who were 50 years or older with at least one additional cardiovascular risk factor and on a stable dose of methotrexate, patients were treated with tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily or a TNF inhibitor. Notably, in February 2019, the dose of tofacitinib in the 10 mg twice daily arm of the study was reduced to 5 mg twice daily after it was determined that the frequency of pulmonary embolism was increased in the tofacitinib 10 mg twice daily treatment arm versus the TNF inhibitor. Additionally, all-cause mortality was increased in the tofacitinib 10 mg twice daily treatment arm versus the TNF inhibitor and tofacitinib 5 mg twice daily treatment arms. In the final study data, patients in the tofacitinib 10 mg twice daily treatment arm were analyzed in their originally randomized treatment group. Results from final safety data from the study for selected events stated as follows.
Mortality: The IRs (95% CI) for all-cause mortality for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, all tofacitinib (combines 5 mg twice daily and 10 mg twice daily treatment arms), and TNF inhibitor were 0.50 (0.33, 0.74), 0.80 (0.57, 1.09), 0.65 (0.50, 0.82), and 0.34 (0.20, 0.54) events per 100 PYs, respectively. Compared with TNF inhibitor, the hazard ratio (HR) (95% CI) for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and all tofacitinib were 1.49 (0.81, 2.74), 2.37 (1.34, 4.18), and 1.91 (1.12, 3.27), respectively.
The IRs (95% CI) for deaths associated with infection for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, all tofacitinib (combines 5 mg twice daily and 10 mg twice daily treatment arms), and TNF inhibitor were 0.08 (0.02, 0.20), 0.18 (0.08, 0.35), 0.13 (0.07, 0.22), and 0.06 (0.01, 0.17) events per 100 PYs, respectively. Compared with TNF inhibitor, the hazard ratio (HR) (95% CI) for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and all tofacitinib were 1.30 (0.29, 5.79), 3.10 (0.84, 11.45), and 2.17 (0.62, 7.62), respectively.
The IRs (95% CI) for deaths associated with cardiovascular events for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, all tofacitinib (combines 5 mg twice daily and 10 mg twice daily treatment arms), and TNF inhibitor were 0.25 (0.13, 0.43), 0.41 (0.25, 0.63), 0.33 (0.23, 0.46), and 0.20 (0.10, 0.36) events per 100 PYs, respectively. Compared with TNF inhibitor, the hazard ratio (HR) (95% CI) for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and all tofacitinib were 1.26 (0.55, 2.88), 2.05 (0.96, 4.39), and 1.65 (0.81, 3.34), respectively.
The IRs (95% CI) for deaths associated with malignancies for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, all tofacitinib (combines 5 mg twice daily and 10 mg twice daily treatment arms), and TNF inhibitor were 0.10 (0.03, 0.23), 0.00 (0.00, 0.08), 0.05 (0.02, 0.12), and 0.02 (0.00, 0.11) events per 100 PYs, respectively. Compared with TNF inhibitor, the hazard ratio (HR) (95% CI) for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and all tofacitinib were 4.88 (0.57, 41.74), 0 (0.00, Inf), and 2.53 (0.30, 21.64), respectively.
The IRs (95% CI) for deaths associated with other causes (excluding infections, cardiovascular events, malignancies) for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, all tofacitinib (combines 5 mg twice daily and 10 mg twice daily treatment arms), and TNF inhibitor were 0.08 (0.02, 0.20), 0.21 (0.10, 0.38), 0.14 (0.08, 0.23), and 0.06 (0.01, 0.17) events per 100 PYs, respectively. Compared with TNF inhibitor, the hazard ratio (HR) (95% CI) for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and all tofacitinib were 1.30 (0.29, 5.81), 3.45 (0.95, 12.54), and 2.34 (0.67, 8.16), respectively.
Infections: The IRs (95% CI) for all infections for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, all tofacitinib (combines 5 mg twice daily and 10 mg twice daily treatment arms), and TNF inhibitor were 41.74 (39.21, 44.39), 48.73 (45.82, 51.77), 45.02 (43.10, 47.01), and 34.24 (32.07, 36.53) patients with events per 100 PYs, respectively. Compared with TNF inhibitor, the hazard ratio (HR) (95% CI) for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and all tofacitinib were 1.20 (1.10, 1.31), 1.36 (1.24, 1.49), and 1.28 (1.18, 1.38), respectively.
The IRs (95% CI) for serious infections for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, all tofacitinib (combines 5 mg twice daily and 10 mg twice daily treatment arms), and TNF inhibitor were 2.86 (2.41, 3.37), 3.64 (3.11, 4.23), 3.24 (2.89, 3.62), and 2.44 (2.02, 2.92) patients with events per 100 PYs, respectively. Compared with TNF inhibitor, the hazard ratio (HR) (95% CI) for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and all tofacitinib were 1.17 (0.92, 1.50), 1.48 (1.17, 1.87), and 1.32 (1.07, 1.63), respectively.
The IRs (95% CI) for opportunistic infections for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, all tofacitinib (combines 5 mg twice daily and 10 mg twice daily treatment arms), and TNF inhibitor were 0.76 (0.54, 1.04), 0.91 (0.66, 1.22), 0.84 (0.67, 1.04), and 0.42 (0.26, 0.64) patients with events per 100 PYs, respectively. Compared with TNF inhibitor, the HRs (95% CI) for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and all tofacitinib were 1.82 (1.07, 3.09), 2.17 (1.29, 3.66), and 1.99 (1.23, 3.22), respectively. The majority of the opportunistic infections in the tofacitinib treatment arms were opportunistic herpes zoster infections; a limited number of events with tuberculosis were also reported. Excluding opportunistic herpes zoster infections and tuberculosis, the IRs (95% CI) for all other opportunistic infections for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, all tofacitinib (combines 5 mg twice daily and 10 mg twice daily treatment arms), and TNF inhibitor were 0.08 (0.02, 0.20), 0.14 (0.06, 0.30), 0.11 (0.05, 0.20), and 0.06 (0.01, 0.17) patients with events per 100 PYs, respectively. Compared with TNF inhibitor, the hazard ratio (HR) (95% CI) for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and all tofacitinib were 1.30 (0.29, 5.82), 2.40 (0.62, 9.29), and 1.84 (0.51, 6.59), respectively.
The IRs (95% CI) for herpes zoster (includes all herpes zoster events) for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, all tofacitinib (combines 5 mg twice daily and 10 mg twice daily treatment arms) and TNF inhibitor were 3.75 (3.22, 4.34), 3.94 (3.38, 4.57), 3.84 (3.45, 4.26), and 1.18 (0.90, 1.52) patients with events per 100 PYs, respectively. Compared with TNF inhibitor, the HR (95% CI) for herpes zoster with tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and all tofacitinib were 3.17 (2.36, 4.27), 3.33 (2.48, 4.48), and 3.25 (2.46, 4.29), respectively.
Serious Infections from Non-interventional Post Approval Safety Study: Data from a non-interventional post approval safety study that evaluated tofacitinib in RA patients from a registry (US Corrona) showed that a numerically higher incidence rate of serious infection was observed for the 11 mg prolonged-release tablet administered once daily than the 5 mg film-coated tablet administered twice daily. Crude incidence rates (95% CI) (i.e., not adjusted for age or sex) from availability of each formulation at 12 months following initiation of treatment were 3.45 (1.93, 5.69) and 2.78 (1.74, 4.21) and at 36 months were 4.71 (3.08, 6.91) and 2.79 (2.01, 3.77) patients with events per 100 patient years in the 11 mg prolonged-release tablet once daily and 5 mg film-coated tablet twice daily groups, respectively. The unadjusted hazard ratio was 1.30 (95% CI: 0.67, 2.50) at 12 months and 1.93 (95% CI: 1.15, 3.24) at 36 months for the 11 mg prolonged-release once daily dose compared to the 5 mg film-coated twice daily dose. Data is based on a small number of patients with events observed with relatively large confidence intervals and limited follow up time available in the 11 mg prolonged-release once daily dose group after 24 months.
Thromboembolism: Venous Thromboembolism: The IRs (95% CI) for VTE for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, all tofacitinib (combines 5 mg twice daily and 10 mg twice daily treatment arms) and TNF inhibitor were 0.33 (0.19, 0.53), 0.70 (0.49, 0.99), 0.51 (0.38, 0.67), and 0.20 (0.10, 0.37) patients with events per 100 PYs, respectively. Compared with TNF inhibitor, the HR (95% CI) for VTE with tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and all tofacitinib were 1.66 (0.76, 3.63), 3.52 (1.74, 7.12), and 2.56 (1.30, 5.05), respectively.
The IRs (95% CI) for PE for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, all tofacitinib (combines 5 mg twice daily and 10 mg twice daily treatment arms) and TNF inhibitor were 0.17 (0.08, 0.33), 0.50 (0.32, 0.74), 0.33 (0.23, 0.46), and 0.06 (0.01, 0.17) patients with events per 100 PYs, respectively. Compared with TNF inhibitor, the HR (95% CI) for PE with tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and all tofacitinib were 2.93 (0.79, 10.83), 8.26 (2.49, 27.43), and 5.53 (1.70, 18.02), respectively. In tofacitinib-treated patients where PE was observed, the majority (97%) had VTE risk factors.
The IRs (95% CI) for DVT for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, all tofacitinib (combines 5 mg twice daily and 10 mg twice daily treatment arms) and TNF inhibitor were 0.21 (0.11, 0.38), 0.31 (0.17, 0.51), 0.26 (0.17, 0.38), and 0.14 (0.06, 0.29) patients with events per 100 PYs, respectively. Compared with TNF inhibitor, the HR (95% CI) for DVT with tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and all tofacitinib were 1.54 (0.60, 3.97), 2.21 (0.90, 5.43), and 1.87 (0.81, 4.30), respectively.
In a post hoc exploratory biomarker analysis within a large randomized PASS in RA patients who were 50 years or older with at least one additional cardiovascular risk factor, occurrences of subsequent VTEs were observed more frequently in tofacitinib-treated patients with D-dimer level ≥2× ULN at 12 months treatment versus those with D-dimer level <2× ULN. This observation was not identified in TNFi-treated patients. Interpretation is limited by the low number of VTE events and restricted D-dimer test availability (only assessed at Baseline, Month 12, and at the end of the study). In patients who did not have a VTE during the study, mean D-dimer levels were significantly reduced at Month 12 relative to Baseline across all treatment arms. However, D-dimer levels ≥2× ULN at Month 12 were observed in approximately 30% of patients without subsequent VTE events, indicating limited specificity of D-dimer testing in this study. Considering the data and the overall limitations of this post hoc exploratory biomarker analysis, there is limited utility of conducting D-dimer monitoring in the context of risk mitigation for VTE events.
Arterial Thromboembolism: The IRs (95% CI) for arterial thromboembolism (ATE) for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, all tofacitinib (combines 5 mg twice daily and 10 mg twice daily treatment arms) and TNF inhibitor were 0.92 (0.68, 1.22), 0.94 (0.68, 1.25), 0.93 (0.75, 1.14), and 0.82 (0.59, 1.12) patients with events per 100 PYs, respectively. Compared with TNF inhibitor, the HR (95% CI) for ATE with tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and all tofacitinib were 1.12 (0.74, 1.70), 1.14 (0.75, 1.74), and 1.13 (0.78, 1.63), respectively.
Major Adverse Cardiovascular Events (MACE), Including Myocardial Infarction: MACE includes non-fatal myocardial infarction, non-fatal stroke, and cardiovascular deaths excluding fatal pulmonary embolism. The IRs (95% CI) for MACE for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, all tofacitinib (combines 5 mg twice daily and 10 mg twice daily treatment arms), and TNF inhibitor were 0.91 (0.67, 1.21), 1.05 (0.78, 1.38), 0.98 (0.79, 1.19), and 0.73 (0.52, 1.01) patients with events per 100 PYs, respectively. Compared with TNF inhibitor, the HRs (95% CI) for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and all tofacitinib were 1.24 (0.81, 1.91), 1.43 (0.94, 2.18), and 1.33 (0.91, 1.94), respectively.
In the tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, all tofacitinib, and TNFi treatment arms, there were a total of 19, 19, 38, and 11 patients with MI events, respectively. Of these totals, the number of patients with fatal MI events was 0, 3, 3, and 3, respectively, whereas the number of patients with non-fatal MI events was 19, 16, 35, and 8, respectively. Therefore, the IRs that follow are for non-fatal MI. The IRs (95% CI) for non-fatal MI for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, all tofacitinib (combines 5 mg twice daily and 10 mg twice daily treatment arms), and TNF inhibitor were 0.37 (0.22, 0.57), 0.33 (0.19, 0.53), 0.35 (0.24, 0.48), and 0.16 (0.07, 0.31) patients with events per 100 PYs, respectively. Compared with TNF inhibitor, the HRs (95% CI) for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and all tofacitinib were 2.32 (1.02, 5.30), 2.08 (0.89, 4.86), and 2.20 (1.02, 4.75), respectively.
Malignancies Excluding NMSC: The IRs (95% CI) for malignancies excluding NMSC for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, all tofacitinib (combines 5 mg twice daily and 10 mg twice daily treatment arms), and TNF inhibitor were 1.13 (0.87, 1.45), 1.13 (0.86, 1.45), 1.13 (0.94, 1.35), and 0.77 (0.55, 1.04) patients with events per 100 PYs, respectively. Compared with TNF inhibitor, the HRs (95% CI) for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and all tofacitinib were 1.47 (1.00, 2.18), 1.48 (1.00, 2.19), and 1.48 (1.04, 2.09), respectively.
The IRs (95% CI) for lymphoma for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, all tofacitinib (combines 5 mg twice daily and 10 mg twice daily treatment arms), and TNF inhibitor were 0.07 (0.02, 0.18), 0.11 (0.04, 0.24), 0.09 (0.04, 0.17), and 0.02 (0.00, 0.10) patients with events per 100 PYs, respectively. Compared with TNF inhibitor, the HRs (95% CI) for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and all tofacitinib were 3.99 (0.45, 35.70), 6.24 (0.75, 51.86), and 5.09 (0.65, 39.78), respectively.
The IRs (95% CI) for lung cancer for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, all tofacitinib (combines 5 mg twice daily and 10 mg twice daily treatment arms), and TNF inhibitor were 0.23 (0.12, 0.40), 0.32 (0.18, 0.51), 0.28 (0.19, 0.39), and 0.13 (0.05, 0.26) patients with events per 100 PYs, respectively. Compared with TNF inhibitor, the HRs (95% CI) for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and all tofacitinib were 1.84 (0.74, 4.62), 2.50 (1.04, 6.02), and 2.17 (0.95, 4.93), respectively.
NMSC: The IRs (95% CI) for NMSC for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, all tofacitinib (combines 5 mg twice daily and 10 mg twice daily treatment arms), and TNF inhibitor were 0.61 (0.41, 0.86), 0.69 (0.47, 0.96), 0.64 (0.50, 0.82), and 0.32 (0.18, 0.52) patients with events per 100 PYs, respectively. Compared with TNF inhibitor, the HRs (95% CI) for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and all tofacitinib were 1.90 (1.04, 3.47), 2.16 (1.19, 3.92), and 2.02 (1.17, 3.50), respectively.
The IRs (95% CI) for basal cell carcinoma for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, all tofacitinib (combines 5 mg twice daily and 10 mg twice daily treatment arms), and TNF inhibitor were 0.37 (0.22, 0.58), 0.33 (0.19, 0.54), 0.35 (0.24, 0.49), and 0.26 (0.14, 0.44) patients with events per 100 PYs, respectively. Compared with TNF inhibitor, the HRs (95% CI) for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and all tofacitinib were 1.43 (0.71, 2.90), 1.28 (0.61, 2.66), and 1.36 (0.72, 2.56), respectively.
The IRs (95% CI) for cutaneous squamous cell carcinoma for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, all tofacitinib (combines 5 mg twice daily and 10 mg twice daily treatment arms), and TNF inhibitor were 0.29 (0.16, 0.48), 0.45 (0.29, 0.69), 0.37 (0.26, 0.51), and 0.16 (0.07, 0.31) patients with events per 100 PYs, respectively. Compared with TNF inhibitor, the HRs (95% CI) for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and all tofacitinib were 1.82 (0.77, 4.30), 2.86 (1.27, 6.43), and 2.32 (1.08, 4.99), respectively.
Gastrointestinal Perforations: The IRs (95% CI) for gastrointestinal perforations for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, all tofacitinib (combines 5 mg twice daily and 10 mg twice daily treatment arms), and TNF inhibitor were 0.17 (0.08, 0.33), 0.10 (0.03, 0.24), 0.14 (0.08, 0.23), and 0.08 (0.02, 0.20) patients with events per 100 PYs, respectively. Compared with TNF inhibitor, the HRs (95% CI) for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and all tofacitinib were 2.20 (0.68, 7.15), 1.29 (0.35, 4.80), and 1.76 (0.58, 5.34), respectively.
Fractures: The IRs (95% CI) for fractures for XELJANZ 5 mg twice daily, XELJANZ 10 mg twice daily, all XELJANZ (combines 5 mg twice daily and 10 mg twice daily treatment arms), and TNF inhibitor were 2.79 (2.34, 3.30), 2.87 (2.40, 3.40), 2.83 (2.50, 3.19) and 2.27 (1.87, 2.74) patients with events per 100 PYs respectively. Compared with TNFi, the HRs (95% CI) for XELJANZ 5 mg twice daily, XELJANZ 10 mg twice daily, and all XELJANZ were 1.23 (0.96, 1.58) 1.26 (0.97, 1.62) and 1.24 (0.99, 1.55) respectively.
Laboratory Tests: Liver enzyme tests: The percentages of patients with at least one post-baseline ALT elevation >1x ULN, 3x ULN, and 5x ULN for the tofacitinib 5 mg twice daily treatment arm were 52.83, 6.01, and 1.68, respectively. The percentages for the tofacitinib 10 mg twice daily treatment arm were 54.46, 6.54, and 1.97, respectively. The percentages for all tofacitinib (combines tofacitinib 5 mg twice daily and tofacitinib 10 mg twice daily) were 53.64, 6.27, and 1.82, respectively. The percentages for the TNF inhibitor treatment arm were 43.33, 3.77, and 1.12, respectively.
The percentages of patients with at least one post-baseline AST elevation >1x ULN, 3x ULN, and 5x ULN for the tofacitinib 5 mg twice daily treatment arm were 45.84, 3.21, and 0.98, respectively. The percentages for the tofacitinib 10 mg twice daily treatment arm were 51.58, 4.57, and 1.62, respectively. The percentages for all tofacitinib (combines tofacitinib 5 mg twice daily and tofacitinib 10 mg twice daily) were 48.70, 3.89, and 1.30, respectively. The percentages for the TNF inhibitor treatment arm were 37.18, 2.38, and 0.70, respectively.
Lipids: At 12 months, in the tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and TNF inhibitor treatment arms, the mean percent increase in LDL cholesterol was 13.80, 17.04, and 5.50, respectively. At 24 months, the mean percent increase was 12.71, 18.14, and 3.64, respectively.
At 12 months, in the tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and TNF inhibitor treatment arms, the mean percent increase in HDL cholesterol was 11.71, 13.63, and 2.82, respectively. At 24 months, the mean percent increase was 11.58, 13.54, and 1.42, respectively.
Clinical Efficacy: Rheumatoid Arthritis: The efficacy and safety of tofacitinib were assessed in five randomized, double-blind, placebo-controlled multicenter studies in patients >18 years with active rheumatoid arthritis diagnosed according to American College of Rheumatology (ACR) criteria. Patients had at least 6 tender and 6 swollen joints at randomization (4 swollen and tender joints for Study II). Tofacitinib, 5 or 10 mg twice daily, was given as monotherapy (Study I) and in combination with DMARDs (Study II) in patients with an inadequate response to those drugs, and in combination with methotrexate in patients with either an inadequate response to MTX (Studies III and Study IV) or inadequate efficacy or lack of tolerance to at least one approved TNF-inhibiting biologic agent (Study V).
Study I was a 6-month monotherapy study in which 610 patients with moderate to severe active rheumatoid arthritis who had an inadequate response to a DMARD (non-biologic or biologic) received tofacitinib 5 or 10 mg twice daily or placebo. At the Month 3 visit, all patients randomized to placebo treatment were advanced in a blinded fashion to a second predetermined treatment of tofacitinib 5 or 10 mg twice daily. The primary endpoints at Month 3 were the proportion of patients who achieved an ACR20 response, changes in Health Assessment Questionnaire - Disability Index (HAQ-DI), and rates of Disease Activity Score DAS28-4(ESR) <2.6.
Study II was a 12-month study in which 792 patients with moderate to severe active rheumatoid arthritis who had an inadequate response to a non-biologic DMARD received tofacitinib 5 or 10 mg twice daily or placebo added to background DMARD treatment (excluding potent immunosuppressive treatments, such as azathioprine or cyclosporine). At the Month 3 visit, non-responding patients randomized to placebo treatment were advanced in a blinded fashion to a second predetermined treatment of tofacitinib 5 or 10 mg twice daily. At the end of Month 6, all placebo patients were advanced to their second predetermined treatment in a blinded fashion. The primary endpoints were the proportion of patients who achieved an ACR20 response at Month 6, changes in HAQ-DI at Month 3 and rates of DAS28-4(ESR) <2.6 at Month 6.
Study III was a 12-month study in which 717 patients with moderate to severe active rheumatoid arthritis who had an inadequate response to MTX. Patients received tofacitinib 5 or 10 mg twice daily, adalimumab 40 mg subcutaneously every other week, or placebo added to background MTX. Placebo patients were advanced as in Study II. The primary endpoints were the proportion of patients who achieved an ACR20 response at Month 6, HAQ-DI at Month 3, and DAS28-4(ESR) less than 2.6 at Month 6. Study III was not designed as a head-to-head comparison between tofacitinib and adalimumab.
Study IV was a 2-year study with a planned analysis at 1 year in which 797 patients with moderate to severe active rheumatoid arthritis who had an inadequate response to MTX received tofacitinib 5 or 10 mg twice daily or placebo added to background MTX. Placebo patients were advanced as in Study II. The primary endpoints were the proportion of patients who achieved an ACR20 response at Month 6, mean change from baseline in van der Heijde-modified total Sharp Score (mTSS) at Month 6, HAQ-DI at Month 3, and DAS28-4(ESR) less than 2.6 at Month 6.
Study V was a 6-month study in which 399 patients with moderate to severe active rheumatoid arthritis who had an inadequate response to at least one approved TNF-inhibiting biologic agent received tofacitinib 5 or 10 mg twice daily or placebo added to background MTX. At the Month 3 visit, all patients randomized to placebo treatment were advanced in a blinded fashion to a second predetermined treatment of tofacitinib 5 or 10 mg twice daily. The primary endpoints at Month 3 were the proportion of patients who achieved an ACR20 response, HAQ-DI, and DAS28-4(ESR) <2.6.
Clinical Response:
ACR Response: The percentages of tofacitinib-treated patients achieving ACR20, ACR50, and ACR70 responses in Studies I, II, IV, and V are shown in Table 1. In all studies, patients treated with either 5 or 10 mg twice daily tofacitinib had statistically significant ACR20, ACR50, and ACR70 response rates at Month 3 and Month 6 vs. placebo treated patients.
In Study IV, ACR20/50/70 response rates at Month 12 were maintained through Month 24.
In Studies I, II, and V, improvement in ACR20 response rate vs. placebo was observed within 2 weeks.
During the 3 month (Studies I and V) and 6 month (Studies II, III, and IV) controlled portions of the studies, patients treated with tofacitinib at a dose of 10 mg twice daily generally had higher response rates compared to patients treated with tofacitinib 5 mg twice daily. In Study III, the primary endpoints were the proportion achieving an ACR20 response at Month 6; change in HAQ-DI at Month 3, and DAS28-4(ESR) <2.6 at Month 6. The data for these primary outcomes were 51.5, 52.6, 47.2 and 28.3%; -0.55, -0.61, -0.49 and -0.24; and 6.2%, 12.5%, 6.7% and 1.1% for the 5 mg twice daily tofacitinib, 10 mg twice daily tofacitinib, adalimumab 40 mg subcutaneously every other week and placebo groups, respectively. For a pre-specified secondary endpoint, the ACR70 response rates at Month 6 for the 5 mg twice daily and 10 mg twice daily tofacitinib groups were significantly greater than adalimumab 19.9%, 21.9% and 9.1%, respectively.
The treatment effect was similar in patients independent of rheumatoid factor status, age, gender, race, or disease status. Time to onset was rapid (as early as Week 2 in Studies I, II and V) and the magnitude of response continued to improve with duration of treatment. As with the overall ACR response in patients treated with 5 mg or 10 mg twice daily tofacitinib, each of the components of the ACR response was consistently improved from baseline including: tender and swollen joint counts; patient and physician global assessment; disability index scores; pain assessment and CRP compared to patients receiving placebo plus MTX or other DMARDs in all studies.
DAS28-4(ESR) Response: Patients in the Phase 3 studies had a mean Disease Activity Score (DAS28-4[ESR]) of 6.1-6.7 at baseline. Significant reductions in DAS28-4(ESR) from baseline (mean improvement) of 1.8-2.0 and 1.9-2.2 were observed in 5 mg and 10 mg tofacitinib-treated patients, respectively, compared to placebo-treated patients (0.7-1.1) at 3 months. The proportion of patients achieving a DAS28 clinical remission (DAS28-4(ESR) <2.6) in Studies II, III and IV was significantly higher in patients receiving 5 mg or 10 mg tofacitinib (6-9% and 13-16%, respectively) compared to 1-3% of placebo patients at 6 months. In Study III, the percentages of patients achieving DAS28-4(ESR) <2.6 observed for tofacitinib 5 mg twice daily, 10 mg twice daily, and adalimumab at Month 6 were 6.2%, 12.5%, and 6.7%, respectively.
In a pooled analysis of the Phase 3 studies, the 10 mg twice daily dose provided increased benefit over the 5 mg twice daily dose in multiple measures of signs and symptoms: improvement from baseline (ACR20, ACR50, and ACR70 response rates), and achievement of targeted disease activity state (either DAS28-4(ESR) <2.6 or ≤3.2). Greater benefits of 10 mg versus 5 mg were shown in the more stringent measures (i.e., ACR70 and DAS28-4(ESR) <2.6 response rates). (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The results of the proportion of patients with an ACR Response for Studies I, II, IV, and V are shown in Table 1. Similar results were observed in Study III.
The results of the components of the ACR response criteria for Study IV and V are shown in Table 2. Similar results were observed for tofacitinib in Studies I, II, III. (See Table 2.)
Click on icon to see table/diagram/image
The percent of ACR20 responders by visit for Study IV is shown in Figure 1. Similar responses were observed for tofacitinib in Studies I, II, III and V. (See Figure 1.)
Click on icon to see table/diagram/image
Physical Function Response and Health-related Outcomes:
Improvement in physical functioning was measured by the HAQ-DI. Patients receiving tofacitinib 5 and 10 mg twice daily demonstrated significantly greater improvement from baseline in physical functioning compared to placebo at Month 3 (Studies I, II, III, and V) and Month 6 (Studies II and III). Tofacitinib 5 or 10 mg twice daily-treated patients exhibited significantly greater improved physical functioning compared to placebo as early as Week 2 in Studies I and II. In Study III, mean HAQ-DI improvements were maintained to 12 months in tofacitinib-treated patients. Mean HAQ-DI improvements were maintained for 36 months in the ongoing open-label extension studies. Compared with adalimumab-treated patients, at Month 3, patients in the tofacitinib 5 mg twice daily had similar decreases from baseline in HAQ-DI values and patients in 10 mg twice daily group had significantly greater decreases in HAQ-DI. The mean change in HAQ-DI from baseline to Month 3 in Studies I to V are shown in Table 3. (See Table 3.)
Click on icon to see table/diagram/image
Other Health-related Outcomes: Health-related quality of life was assessed by the Short Form Health Survey (SF-36) in all 5 studies. In these studies, patients receiving tofacitinib 10 mg twice daily demonstrated significantly greater improvement from baseline compared to placebo in physical component summary (PCS), mental component summary (MCS) scores and in all 8 domains of the SF-36 at Month 3. Both tofacitinib-treated groups exhibited significantly greater improvement from baseline compared to placebo in all 8 domains as well as PCS and MCS at Month 3 in Studies I, IV, and V. In Studies III and IV, mean SF-36 improvements were maintained to 12 months in tofacitinib-treated patients.
Improvement in fatigue was evaluated by the Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-F) scale at Month 3 in all studies. Patients receiving tofacitinib 5 or 10 mg twice daily demonstrated significantly greater improvement from baseline in fatigue compared to placebo in all 5 studies. In Studies III and IV, mean FACIT-F improvements were maintained to 12 months in tofacitinib-treated patients.
Improvement in sleep was assessed using the Sleep Problems Index I and II summary scales of the Medical Outcomes Study Sleep (MOS-Sleep) measure at Month 3 in all studies. Patients receiving tofacitinib 5 or 10 mg twice daily demonstrated significantly greater improvement from baseline in both scales compared to placebo in Studies II, III, and IV. In Studies III and IV, mean improvements in both scales were maintained to 12 months in tofacitinib-treated patients.
Improvement in productivity was evaluated using the Work Limitations Questionnaire (WLQ) scale at Month 3 in all studies. Patients receiving tofacitinib 10 mg twice daily demonstrated significantly greater improvement from baseline in the Overall Output Summary Scale compared to placebo in Studies III, IV, and V. In Studies III and IV, mean Overall Output improvements were maintained to 12 months in tofacitinib 10 mg twice daily-treated patients.
Durability of Clinical Responses:
Durability of effect was assessed by ACR20, ACR50, ACR70 response rates, mean HAQ-DI, and mean DAS28-4(ESR) in the three Phase 3 DMARD IR studies with duration of at least one year. Efficacy was maintained in all tofacitinib treatment groups through to the end of the studies. Evidence of persistence of efficacy with tofacitinib treatment for up to 6 years is also provided from data in a large randomized PASS in RA patients 50 years and older with at least one additional CV risk factor, as well as in completed open-label, long-term follow-up studies up to 8 years.
Psoriatic Arthritis: The efficacy and safety of tofacitinib were assessed in 2 randomized, double-blind, placebo-controlled Phase 3 studies in adult patients with active PsA (≥3 swollen and ≥3 tender joints). Patients were required to have active plaque psoriasis at the screening visit. For both studies, the primary endpoints were ACR20 response rate and change from baseline in HAQ-DI at Month 3.
Study PsA-I (OPAL BROADEN) evaluated 422 patients who had a previous inadequate response (due to lack of efficacy or intolerance) to a csDMARD (MTX for 92.7% of patients); 32.7% of the patients in this study had a previous inadequate response to >1 csDMARD or 1 csDMARD and a targeted synthetic DMARD (tsDMARD). In OPAL BROADEN, previous treatment with TNF inhibitor was not allowed. All patients were required to have 1 concomitant csDMARD; 83.9% of patients received concomitant MTX, 9.5% of patients received concomitant sulfasalazine, and 5.7% of patients received concomitant leflunomide. The median PsA disease duration was 3.8 years. At baseline, 79.9% and 56.2% of patients had enthesitis and dactylitis, respectively. Patients randomized to tofacitinib received 5 mg twice daily or tofacitinib 10 mg twice daily for 12 months. Patients randomized to placebo were advanced in a blinded manner at Month 3 to either tofacitinib 5 mg twice daily or tofacitinib 10 mg twice daily and received treatment until Month 12. Patients randomized to adalimumab (active control arm) received 40 mg subcutaneously every 2 weeks for 12 months.
Study PsA-II (OPAL BEYOND) evaluated 394 patients who had discontinued a TNF inhibitor due to lack of efficacy or intolerance; 36.0% had a previous inadequate response to >1 biological DMARD. All patients were required to have 1 concomitant csDMARD; 71.6% of patients received concomitant MTX, 15.7% of patients received concomitant sulfasalazine, and 8.6% of patients received concomitant leflunomide. The median PsA disease duration was 7.5 years. At baseline, 80.7% and 49.2% of patients had enthesitis and dactylitis, respectively. Patients randomized to tofacitinib received 5 mg twice daily or tofacitinib 10 mg twice daily for 6 months. Patients randomized to placebo were advanced in a blinded manner at Month 3 to either tofacitinib 5 mg twice daily or tofacitinib 10 mg twice daily and received treatment until Month 6.
Signs and Symptoms: Treatment with tofacitinib resulted in significant improvements in some signs and symptoms of PsA, as assessed by the ACR20 response criteria compared to placebo at Month 3. The efficacy results for important endpoints assessed are shown in Table 4. (See Table 4.)
Click on icon to see table/diagram/image
Both TNF inhibitor-naïve and TNF inhibitor inadequate responder tofacitinib 5 mg BID-treated patients had significantly higher ACR20 response rates compared to placebo at Month 3. Examination of age, sex, race, baseline disease activity and PsA subtype did not identify differences in response to tofacitinib. The number of patients with arthritis mutilans or axial involvement was too small to allow meaningful assessment. Statistically significant ACR20 response rates were observed with tofacitinib 5 mg BID in both studies as early as Week 2 (first post-baseline assessment) in comparison to placebo.
In OPAL BROADEN, Minimal Disease Activity (MDA) response was achieved by 26.2%, 25.5% and 6.7% of tofacitinib 5 mg BID, adalimumab and placebo treated patients, respectively (tofacitinib 5 mg BID treatment difference from placebo 19.5% [95% CI: 9.9, 29.1]) at Month 3. In OPAL BEYOND, MDA was achieved by 22.9% and 14.5% of tofacitinib 5 mg BID and placebo treated patients, respectively, however tofacitinib 5 mg BID did not achieve nominal statistical significance (treatment difference from placebo 8.4% [95% CI: -1.0, 17.8] at Month 3).
Radiographic Response: In study OPAL BROADEN, the progression of structural joint damage was assessed radiographically utilizing the van der Heijde-modified Total Sharp Score (mTSS) and the proportion of patients with radiographic progression (mTSS increase from baseline greater than 0.5) was assessed at Month 12. At Month 12, 96% and 98% of patients receiving tofacitinib 5 mg twice daily, and adalimumab 40 mg subcutaneously every 2 weeks, respectively, did not have radiographic progression (mTSS increase from baseline less than or equal to 0.5).
Physical Function and Health-related Quality of Life: Improvement in physical functioning was measured by the HAQ-DI. Patients receiving tofacitinib 5 mg twice daily demonstrated greater improvement (p≤0.05) from baseline in physical functioning compared to placebo at Month 3 (see Table 5).
Click on icon to see table/diagram/image
The HAQ-DI responder rate (response defined as having decrease from baseline of ≥0.35) at month 3 in studies OPAL BROADEN and OPAL BEYOND was 53% and 50%, respectively in patients receiving tofacitinib 5 mg twice daily, 31% and 28%, respectively in patients receiving placebo, and 53% in patients receiving adalimumab 40 mg subcutaneously once every 2 weeks (OPAL BROADEN only).
Health-related quality of life was assessed by SF-36v2, fatigue was assessed by the FACIT-F. Patients receiving tofacitinib 5 mg twice daily demonstrated greater improvement from baseline compared to placebo in the SF-36v2 physical functioning domain, the SF-36v2 physical component summary score, and FACIT-F scores at month 3 in studies OPAL BROADEN and OPAL BEYOND (nominal p≤0.05). Improvements from baseline in SF-36v2 and FACIT-F were maintained through Month 6 (OPAL BROADEN and OPAL BEYOND) and Month 12 (OPAL BROADEN).
Patients receiving tofacitinib 5 mg twice daily demonstrated a greater improvement in arthritis pain (as measured on a 0-100 visual analogue scale) from baseline at Week 2 (first post-baseline assessment) through Month 3 compared to placebo in studies OPAL BROADEN and OPAL BEYOND (nominal p≤0.05).
Ulcerative Colitis: The efficacy and safety of tofacitinib for the treatment of adult patients with moderately to severely active UC (Mayo score 6 to 12 with endoscopy subscore ≥2 and rectal bleeding subscore ≥1) were assessed in 3 multicenter, double-blind, randomized, placebo-controlled studies: 2 identical induction studies (OCTAVE Induction 1 and OCTAVE Induction 2) followed by 1 maintenance study (OCTAVE Sustain). Enrolled patients had failed at least 1 conventional therapy, including corticosteroids, immunomodulators, and/or a TNF inhibitor. Concomitant stable doses of oral aminosalicylates and corticosteroids (prednisone or equivalent daily dose up to 25 mg) were permitted with taper of corticosteroids to discontinuation mandated within 15 weeks of entering the maintenance study. Tofacitinib was administered as monotherapy (i.e., without concomitant use of biologics and immunosuppressants) for UC.
Table 6 provides additional information regarding pertinent study design and population characteristics. (See Table 6.)
Click on icon to see table/diagram/image
In addition, safety and efficacy of tofacitinib were assessed in an open-label long-term extension study (OCTAVE Open). Patients who completed 1 of the induction studies (OCTAVE Induction 1 or OCTAVE Induction 2) but did not achieve clinical response or patients who completed or withdrew early due to treatment failure in the maintenance study (OCTAVE Sustain) were eligible for OCTAVE Open. Patients from OCTAVE Induction 1 or OCTAVE Induction 2 who did not achieve clinical response after 8 weeks in OCTAVE Open were to be discontinued from OCTAVE Open. Corticosteroid tapering was also required upon entrance into OCTAVE Open.
Induction Efficacy Data (OCTAVE Induction 1 and OCTAVE Induction 2): The primary endpoint of OCTAVE Induction 1 and OCTAVE Induction 2 was the proportion of patients in remission at Week 8, and the key secondary endpoint was the proportion of patients with improvement of endoscopic appearance of the mucosa at Week 8. Remission was defined as clinical remission (a total Mayo score ≤2 with no individual subscore >1) and rectal bleeding subscore of 0. Improvement of endoscopic appearance of the mucosa was defined as endoscopy subscore of 0 or 1.
A significantly greater proportion of patients treated with tofacitinib 10 mg twice daily achieved remission, improvement of endoscopic appearance of the mucosa, and clinical response at Week 8 compared to placebo in both studies, as shown in Table 7.
The efficacy results based on the endoscopic readings at the study sites were consistent with the results based on the central endoscopy readings. (See Table 7.)
Click on icon to see table/diagram/image
In both subgroups of patients with or without prior TNF inhibitor failure, a greater proportion of patients treated with tofacitinib 10 mg twice daily achieved remission and improvement of endoscopic appearance of the mucosa at Week 8 as compared to placebo. This treatment difference was consistent between the 2 subgroups (see Table 8).
Click on icon to see table/diagram/image
As early as Week 2, the earliest scheduled study visit, and at each visit thereafter, significant differences were observed between tofacitinib 10 mg twice daily and placebo in the change from baseline in rectal bleeding and stool frequency, and partial Mayo score.
Maintenance (OCTAVE Sustain): Patients who completed 8 weeks in 1 of the induction studies and achieved clinical response were re randomized into OCTAVE Sustain; 179 out of 593 (30.2%) patients were in remission at baseline of OCTAVE Sustain.
The primary endpoint in OCTAVE Sustain was the proportion of patients in remission at Week 52. The 2 key secondary endpoints were the proportion of patients with improvement of endoscopic appearance at Week 52, and the proportion of patients with sustained corticosteroid free remission at both Week 24 and Week 52 among patients in remission at baseline of OCTAVE Sustain.
A significantly greater proportion of patients in both the tofacitinib 5 mg twice daily and tofacitinib 10 mg twice daily treatment groups achieved the following endpoints at Week 52 as compared to placebo: remission, improvement of endoscopic appearance of the mucosa, normalization of endoscopic appearance of the mucosa, maintenance of clinical response, remission among patients in remission at baseline, and sustained corticosteroid-free remission at both Week 24 and Week 52 among patients in remission at baseline, as shown in Table 9. (See Table 9.)
Click on icon to see table/diagram/image
In both subgroups of patients with or without prior TNF inhibitor failure, a greater proportion of patients treated with either tofacitinib 5 mg twice daily or tofacitinib 10 mg twice daily achieved the following endpoints at Week 52 of OCTAVE Sustain as compared to placebo: remission, improvement of endoscopic appearance of the mucosa, or sustained corticosteroid-free remission at both Week 24 and Week 52 among patients in remission at baseline (Table 10). This treatment difference from placebo was similar between tofacitinib 5 mg twice daily and tofacitinib 10 mg twice daily in the subgroup of patients without prior TNF inhibitor failure. In the subgroup of patients with prior TNF inhibitor failure, the observed treatment difference from placebo was numerically greater for tofacitinib 10 mg twice daily than tofacitinib 5 mg twice daily by 9.7 to 16.7 percentage points across the primary and key secondary endpoints. (See Table 10.)
Click on icon to see table/diagram/image
The proportion of patients in both tofacitinib groups who had treatment failure was lower compared to placebo at each time point as early as Week 8, the first time point where treatment failure was assessed, as shown in Figure 2. (See Figure 2.)
Click on icon to see table/diagram/image
Health-related and Quality of Life Outcomes: Tofacitinib 10 mg twice daily demonstrated greater improvement from baseline compared to placebo in physical component summary (PCS) and mental component summary (MCS) scores, and in all 8 domains of the SF-36 in the induction studies (OCTAVE Induction 1, OCTAVE Induction 2). In the maintenance study (OCTAVE Sustain), tofacitinib 5 mg twice daily or tofacitinib 10 mg twice daily demonstrated greater maintenance of improvement compared to placebo in PCS and MCS scores, and in all 8 domains of the SF-36 at Week 24 and Week 52.
Tofacitinib 10 mg twice daily demonstrated greater improvement from baseline compared to placebo at week 8 in the total and all 4 domain scores of the Inflammatory Bowel Disease Questionnaire (IBDQ) (bowel symptoms, systemic function, emotional function, and social function) in the induction studies (OCTAVE Induction 1, OCTAVE Induction 2). In the maintenance study (OCTAVE Sustain), tofacitinib 5 mg twice daily or tofacitinib 10 mg twice daily demonstrated greater maintenance of improvement compared to placebo in the total and all 4 domain scores of the IBDQ at Week 24 and Week 52.
Improvements were also observed in the EuroQoL 5-Dimension (EQ-5D) and various domains of the Work Productivity and Activity Impairment (WPAI-UC) questionnaire in both induction and maintenance studies compared to placebo.
Open-label Extension Study (OCTAVE Open): Patients who did not achieve clinical response in one of the induction studies (OCTAVE Induction 1 or OCTAVE Induction 2) after 8 weeks of tofacitinib 10 mg twice daily were allowed to enter an open-label extension study (OCTAVE Open). After an additional 8 weeks of tofacitinib 10 mg twice daily in OCTAVE Open, 53% (154/293) patients achieved clinical response and 14% (42/293) patients achieved remission.
Patients who achieved clinical response in 1 of the induction studies (OCTAVE Induction 1 or OCTAVE Induction 2) with tofacitinib 10 mg twice daily but experienced treatment failure after their dose was reduced to tofacitinib 5 mg twice daily or following treatment interruption in OCTAVE Sustain (i.e., were randomized to placebo), had their dose increased to tofacitinib 10 mg twice daily in OCTAVE Open. After 8 weeks on tofacitinib 10 mg twice daily in OCTAVE Open, remission was achieved in 35% (20/58) patients who received tofacitinib 5 mg twice daily in OCTAVE Sustain and 40% (40/99) patients with dose interruption in OCTAVE Sustain. At Month 12 in OCTAVE Open, 52% (25/48) and 45% (37/83) of these patients achieved remission, respectively.
Furthermore, at Month 12 of study OCTAVE Open, 74% (48/65) of patients who achieved remission at the end of study OCTAVE Sustain on either tofacitinib 5 mg twice daily or tofacitinib 10 mg twice daily remained in remission while receiving tofacitinib 5 mg twice daily.
Pharmacokinetics: The PK profile of tofacitinib is characterized by rapid absorption (peak plasma concentrations are reached within 0.5-1 hour), rapid elimination (half-life of ~3 hours) and dose-proportional increases in systemic exposure. Steady-state concentrations are achieved in 24-48 hours with negligible accumulation after twice daily administration.
Absorption and Distribution: Tofacitinib is well-absorbed, with an oral bioavailability of 74%. Co-administration of tofacitinib with a high-fat meal resulted in no changes in AUC while C
max was reduced by 32%. In clinical trials, tofacitinib was administered without regard to meal.
After intravenous administration, the volume of distribution is 87 L. Approximately 40% of circulating tofacitinib is bound to proteins. Tofacitinib binds predominantly to albumin and does not appear to bind to α1-acid glycoprotein. Tofacitinib distributes equally between red blood cells and plasma.
Metabolism and Elimination: Clearance mechanisms for tofacitinib are approximately 70% hepatic metabolism and 30% renal excretion of the parent drug. The metabolism of tofacitinib is primarily mediated by CYP3A4 with minor contribution from CYP2C19. In a human radiolabeled study, more than 65% of the total circulating radioactivity was accounted for by unchanged drug, with the remaining 35% attributed to 8 metabolites, each accounting for less than 8% of total radioactivity. The pharmacologic activity of tofacitinib is attributed to the parent molecule.
In vitro, tofacitinib is a substrate for multidrug resistance (MDR) 1, but not for breast cancer resistance protein (BCRP), organic anion transporting polypeptide (OATP) 1B1/1B3, or organic cationic transporter (OCT) 1/2, and is not an inhibitor of MDR1, OAT P1B1/1B3, OCT2, organic anion transporter (OAT) 1/3, or multidrug resistance associated protein (MRP) at clinically meaningful concentrations.
Pharmacokinetic data and dosing recommendations for special populations and drug interactions are provided in Figure 3.
Modifications required for special populations are described in Dosage & Administration.
Pharmacokinetics in RA Patients: Population PK analysis in rheumatoid arthritis patients indicated that systemic exposure (AUC) of tofacitinib in the extremes of body weight (40 kg, 140 kg) were similar to that of a 70 kg patient. Elderly patients 80 years of age were estimated to have <5% higher AUC relative to the mean age of 55 years. Women were estimated to have 7% lower AUC compared to men. The available data have also shown that there are no major differences in tofacitinib AUC between White, Black and Asian patients. An approximate linear relationship between body weight and volume of distribution was observed, resulting in higher peak (C
max) and lower trough (C
min) concentrations in lighter patients. However, this difference is not considered to be clinically relevant. The between-subject variability (percentage coefficient of variation) in AUC of tofacitinib is estimated to be approximately 27%.
Pharmacokinetics in Patients with Active Psoriatic Arthritis / Moderate to Severe UC: Results from population PK analysis in patients with active PsA or moderate to severe UC were consistent with those in patients with RA.
Renal Impairment: Patients with mild, moderate, and severe renal impairment had 37%, 43%, and 123% higher AUC, respectively, compared with healthy patients (see Dosage & Administration). In patients with end-stage renal disease, the contribution of dialysis to the total clearance of tofacitinib was relatively small.
Hepatic Impairment: Patients with mild and moderate hepatic impairment had 3%, and 65% higher AUC, respectively, compared with healthy patients. Patients with severe hepatic impairment were not studied (see Dosage & Administration).
Pediatric Population: The pharmacokinetics, safety and efficacy of tofacitinib in pediatric patients have not been established. (See Figure 3.)
Click on icon to see table/diagram/image
Reference values for weight, age, gender, and race comparisons are 70 kg, 55 years, male and White, respectively; reference groups for renal and hepatic impairment data are subjects with normal renal or hepatic function.
Toxicology: Non-clinical safety data: In nonclinical studies, effects were observed on the immune and hematopoietic systems that were attributed to the pharmacological properties (JAK inhibition) of tofacitinib. Secondary effects from immunosuppression, such as bacterial and viral infections and lymphoma were observed at clinically relevant doses. Other findings at doses well above human exposures included effects on the liver, lung and gastrointestinal systems.
Lymphoma was observed in 3 of 8 adult and 0 of 14 juvenile monkeys dosed with tofacitinib at 5 mg/kg twice daily. The no observed adverse effect level (NOAEL) for the lymphomas was 1 mg/kg twice daily. The unbound AUC at 1 mg/kg twice daily was 341 ng·h/mL, which is approximately half of the unbound AUC at 10 mg twice daily and similar to the unbound AUC at 5 mg twice daily in humans.
Tofacitinib is not mutagenic or genotoxic based on the results of a series of
in vitro and
in vivo tests for gene mutations and chromosomal aberrations.
The carcinogenic potential of tofacitinib was assessed in 6-month rasH2 transgenic mouse carcinogenicity and 2-year rat carcinogenicity studies. Tofacitinib was not carcinogenic in mice up to a high dose of 200 mg/kg/day (unbound drug AUC of ~19-fold the human AUC at 10 mg twice daily). Benign Leydig cell tumors were observed in rats: benign Leydig cell tumors in rats are not associated with a risk of Leydig cell tumors in humans. Hibernomas (malignancy of brown adipose tissue) were observed in female rats at doses ≥30 mg/kg/day (unbound drug AUC of ~41-fold the human AUC at 10 mg twice daily). Benign thymomas were observed in female rats dosed only at the 100 reduced to 75 mg/kg/day dose (unbound drug AUC of ~94-fold the human AUC at 10 mg twice daily).
Tofacitinib was shown to be teratogenic in rats and rabbits, and have effects in rats on female fertility, parturition, and peri/post-natal development. Tofacitinib had no effects on male fertility, sperm motility, or sperm concentration. Tofacitinib was secreted in milk of lactating rats. In studies conducted in juvenile rats and monkeys tofacitinib-related effects on the immune system were similar to those in adult animals. There were no tofacitinib-related effects on reproductive system or bone development in males or females.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image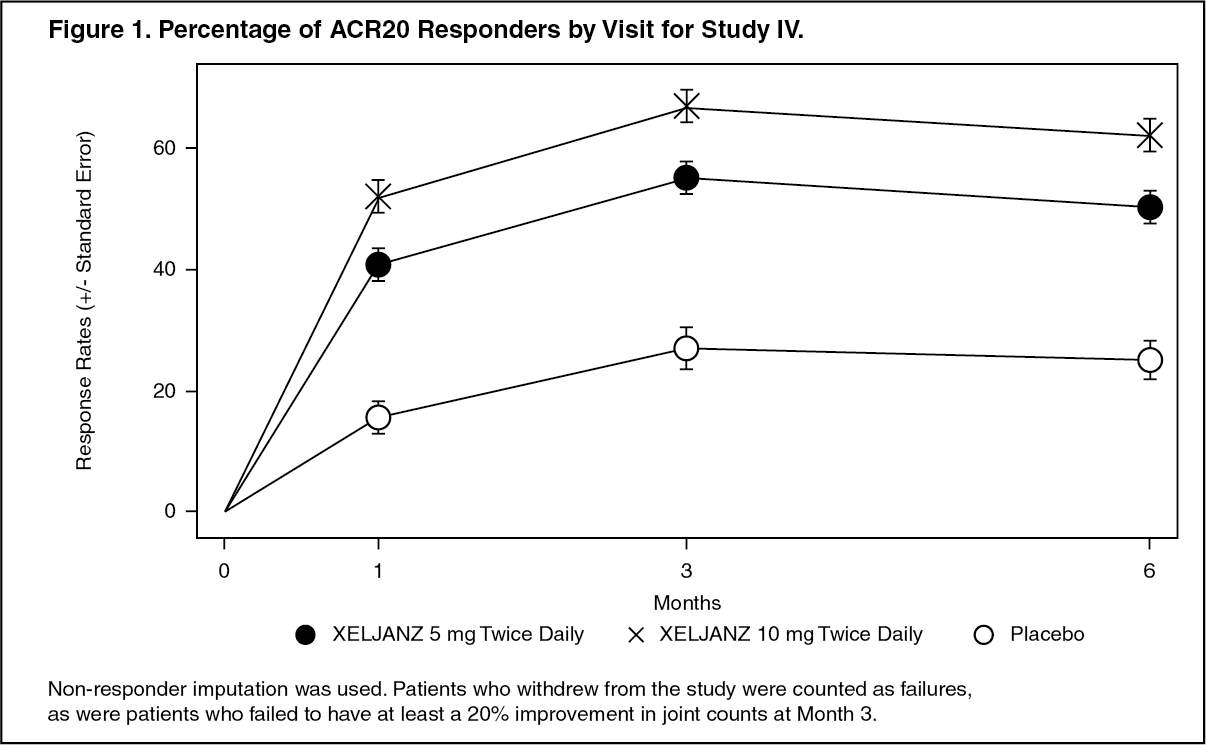 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image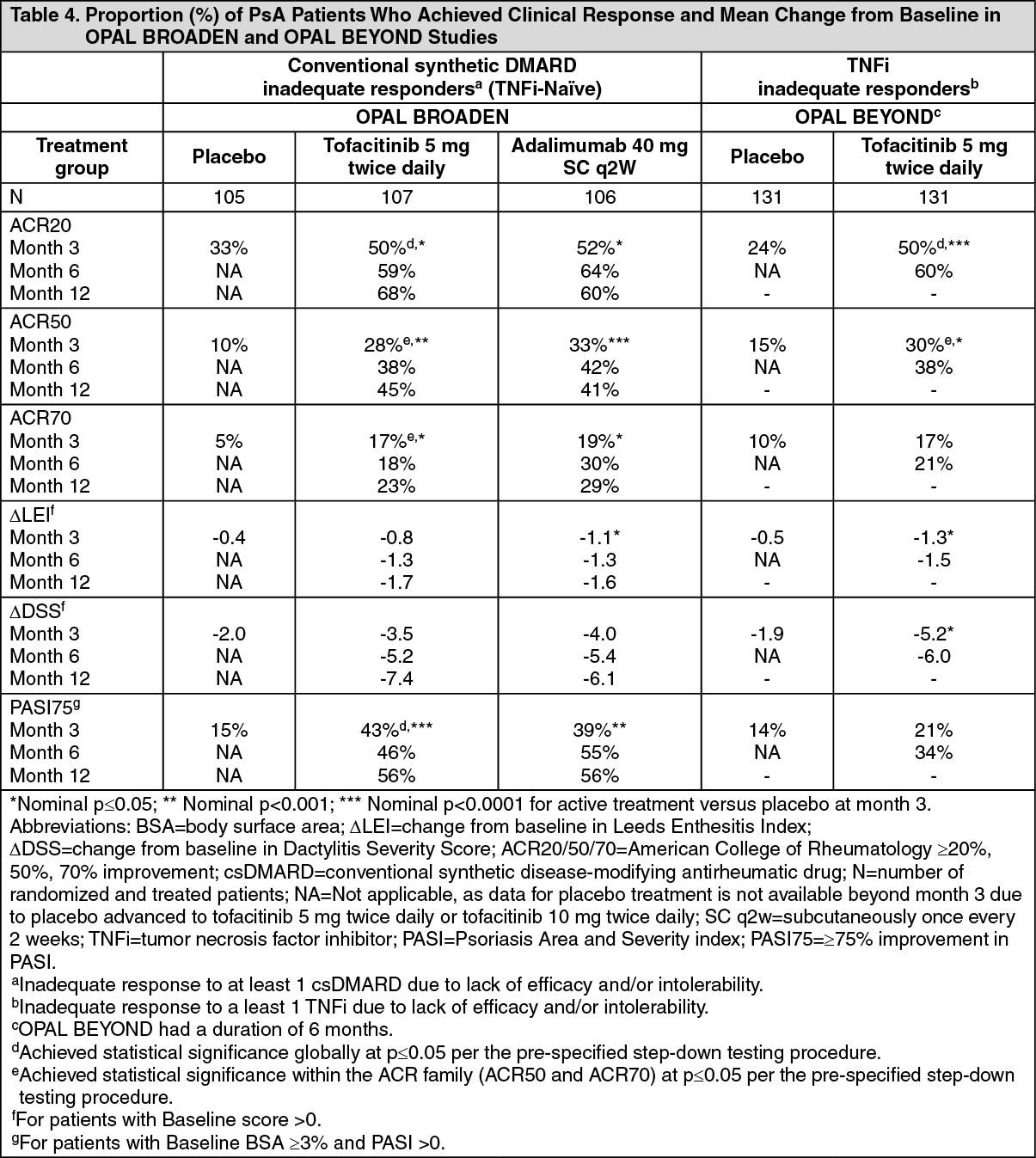 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
