


 Sign Out
Sign Out


Gabapentin binds with high affinity to the α2-δ (alpha-2-delta) subunit of voltage-gated calcium channels. Broad panel screening suggests it does not bind to other neurotransmitter receptors of the brain and does not interact with sodium channels.
The relevance of the binding activity of gabapentin to the anticonvulsant effects in animal models and in humans remains to be established (see DETAILED PHARMACOLOGY as follows).
CLINICAL TRIALS: Comparative Bioavailability Studies: A comparative bioavailability study of pms-GABAPENTIN 400 mg capsules was performed. Pharmacokinetic and bioavailability data were measured in 30 volunteers in the fasting state. The results are summarized as follows in Table 1. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageA single center, randomized, single-dose, double-blinded, 2-period, 2-sequence, crossover, comparative oral bioavailability study was conducted to compare pms-GABAPENTIN (gabapentin) 600 mg tablets of Pharmascience Inc. Canada and NEURONTIN (gabapentin) 600 mg tablets of Warner-Lambert Company LLC Canada Inc. (currently manufactured by Pfizer Canada Inc.) both administered as a 1 x 600 mg dose to 23 healthy male volunteers under fasting conditions. Bioavailability data were measured and the results are summarized in Table 2. (See Table 2.)
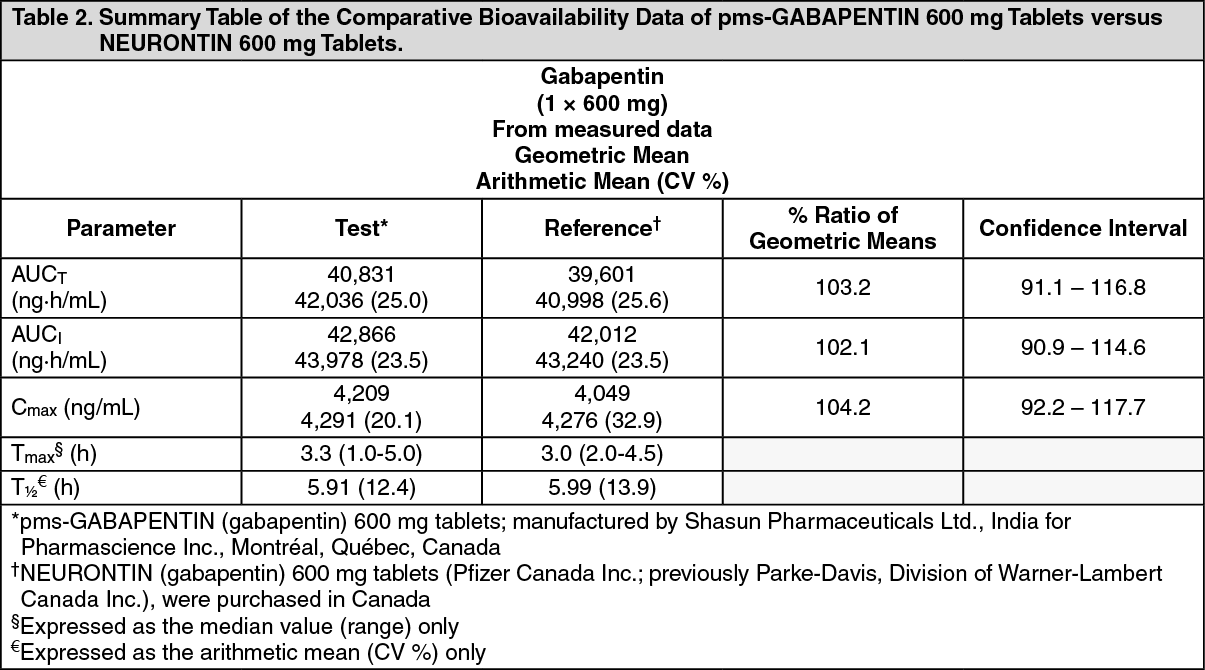 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDETAILED PHARMACOLOGY: Animal Pharmacology: In Vitro Studies: The mechanism of the anticonvulsant action of gabapentin appears to be distinctly different from that of other antiepileptic drugs. Although structurally similar to GABA, gabapentin at concentrations up to 1,000 mcM, did not bind to GABA receptors, it was not metabolized to GABA or a GABA agonist, and it did not inhibit the uptake of GABA or its degradation by GABA-transaminase. Therefore, it does not appear to act through any known GABA mechanism, in contrast to the benzodiazepines, barbiturates, sodium valproate and other similar agents. Gabapentin (0.01-100 mcM) did not interact with neuronal sodium channels or L-type calcium channels, in contrast to phenytoin, carbamazepine and sodium valproate which interact with these to promote the stability of excitable membranes. Finally, gabapentin (0.01-100 mcM) did not interact with glutamate, glycine or N-methyl-D-aspartate (NMDA) receptors, in contrast to other drugs that have demonstrated anticonvulsant activity in animal models following interaction with these receptors. These neurophysiological findings indicate that gabapentin has a mechanism of action different from that of commonly used antiepileptic drugs.
Gabapentin binds with high affinity to the α2-δ (alpha-2-delta) subunit of voltage-gated calcium channels. Auto-radiographic studies have confirmed that there are high levels of gabapentin binding in the outer layers of the cerebral cortex and other regions of the brain with major excitatory input, such as the hippocampus and cerebellum, that are known to be associated with seizure activity.
In Vivo Studies: Gabapentin has been shown to have anticonvulsant activity in animal models typically used to characterize anticonvulsant activity. Gabapentin prevented seizures induced by maximal electroshock in mice and rats in a dose-dependent manner (ED50, 200 mg/kg and 9 mg/kg in mice and rats, respectively). Peak anticonvulsant effects were seen approximately 120-240 minutes post-dose.
Gabapentin prevented threshold clonic convulsions induced by the convulsant pentylenetetrazol in mice (ED50 450 mg/kg); the threshold dose of pentylenetetrazol needed to produce clonic seizures was significantly elevated by gabapentin.
Gabapentin treatment prevented tonic extensor seizures in mice from a variety of convulsant agents, including bicuculline, picrotoxin, strychnine and thiosemicarbazide.
Administration of gabapentin to kindled rats significantly reduced motor seizures from electrical stimulation of the brain, but had relatively little effect on the threshold for electrical after discharges at the site of stimulation.
Experiments with genetically-susceptible animals showed that gabapentin prevented generalized convulsive seizures. However, results with other genetic models indicated that gabapentin would be ineffective against photosensitive myoclonic seizures and absence seizures.
The anticonvulsant effects of gabapentin add to those of several other anticonvulsants against maximal electroshock in mice, thus suggesting that gabapentin would be useful as add-on therapy.
Pharmacokinetics: All pharmacological actions following gabapentin administration are due to the activity of the parent compound; gabapentin is not metabolized to a significant extent in humans.
Plasma gabapentin concentrations are dose-proportional at doses of 300 to 400 mg q8h, ranging between 1 mcg/mL and 10 mcg/mL, but are less than dose-proportional above the clinical range (>600 mg q8h). There is no correlation between plasma levels and efficacy.
Gabapentin pharmacokinetics are not affected by repeated administration, and steady-state plasma concentrations are predictable from single dose data. Gabapentin steady-state pharmacokinetics are similar for healthy subjects and patients with epilepsy receiving antiepileptic agents.
Absorption: Following oral administration of gabapentin, peak plasma concentrations are observed within 2 to 3 hours. Absolute bioavailability of a 300 mg dose of gabapentin capsules is approximately 59%. At doses of 300 and 400 mg, gabapentin bioavailability is unchanged following multiple dose administration.
Food has no effect on the rate or extent of absorption of gabapentin.
Distribution: Less than 3% of gabapentin is bound to plasma proteins. The apparent volume of distribution of gabapentin after 150 mg intravenous administration is 58+6 L (Mean ± SD). In patients with epilepsy, gabapentin concentrations in cerebrospinal fluid are approximately 20% of corresponding steady-state trough plasma concentrations.
Metabolism: Gabapentin is not metabolized to a significant extent in humans. Gabapentin does not induce or inhibit hepatic mixed function oxidase enzymes responsible for drug metabolism and does not interfere with the metabolism of commonly co-administered antiepileptic drugs.
Excretion: Gabapentin is eliminated solely by renal excretion as unchanged drug, and can be removed from plasma by hemodialysis. Gabapentin elimination rate constant, plasma clearance and renal clearance are directly proportional to creatinine clearance. The elimination half-life of gabapentin is independent of dose and averages 5 to 7 hours in subjects with normal renal function.
Table 3 summarizes the mean steady-state pharmacokinetic parameters of gabapentin capsules. (See Table 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageBioequivalence of Dosage Forms: Gabapentin 600 mg and 800 mg tablets are bioequivalent to two 300 mg capsules and two 400 mg capsules, respectively. The results of a single-dose, two-way crossover, comparative bioavailability study in the fasted state comparing gabapentin 600 mg tablets and 2 x 300 mg gabapentin capsules are summarized as follows (Table 4). (See Table 4.)
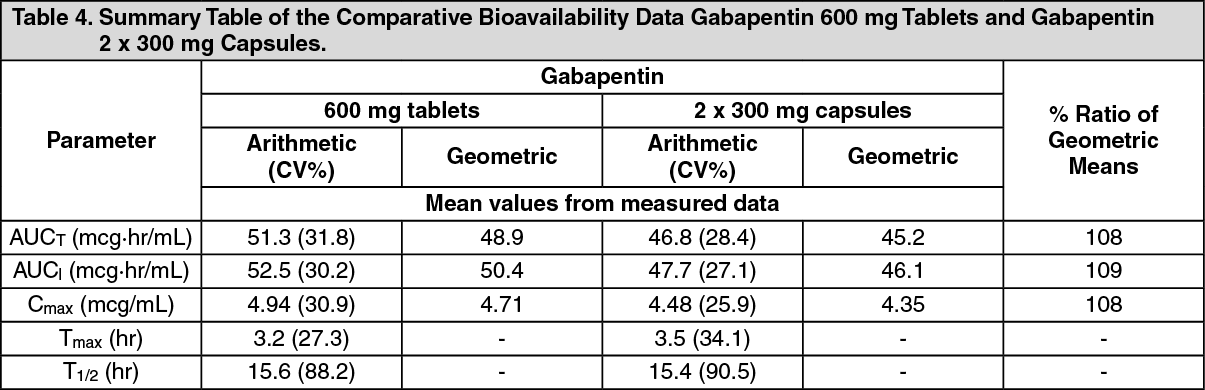 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSpecial Populations and Conditions: Pediatrics: There are no pharmacokinetic data available in children under 18 years of age.
Geriatrics: Apparent oral clearance (CL/F) of gabapentin decreased as age increased, from about 225 mL/min in subjects under 30 years of age to about 125 mL/min in subjects over 70 years of age. Renal clearance (CLr) of gabapentin also declined with age; however, this decrease can largely be explained by the decline in renal function. Reduction of gabapentin dose may be required in patients who have age-related compromised renal function (see Dosing Considerations under DOSAGE & ADMINISTRATION).
Hepatic Insufficiency: Because gabapentin is not metabolized to a significant extent in humans, no study was performed in patients with hepatic impairment.
Renal Insufficiency: In patients with impaired renal function, gabapentin clearance is markedly reduced and dosage adjustment is necessary (see Dosing Considerations and Special Patient Populations: Geriatrics and Renal Impairment (Table 5) under DOSAGE AND ADMINISTRATION).
Hemodialysis: In a study in anuric subjects (N = 11), the apparent elimination half-life of gabapentin on non-dialysis days was about 132 hours; during dialysis the apparent half-life of gabapentin was reduced to 3.8 hours. Hemodialysis thus has a significant effect on gabapentin elimination in anuric subjects.
Dosage adjustment in patients undergoing hemodialysis is necessary (see Dosing Considerations and Special Patient Populations: Geriatrics and Renal Impairment (Table 5) under DOSAGE AND ADMINISTRATION).
Toxicology: Acute Toxicity: Gabapentin exhibited a very low order of acute toxicity in rodents and monkeys. In adult and 3-week-old mice, no deaths occurred and median lethal doses (MLD's) were not identified, being greater than 8,000, 2,000, and 4,000 mg/kg by the oral, intravenous, and subcutaneous routes, respectively. In adult and 3-week-old rats, MLD's after single oral and intravenous doses were greater than 8,000 and 2,000 mg/kg, respectively. No signs of toxicity were noted in monkeys given single oral doses of gabapentin up to 1,250 mg/kg.
Chronic Toxicity: Multi-dose oral administration of gabapentin was well tolerated in all species tested (mice, rats, dogs, monkeys). Decreased body weight gain was observed in rats; hypoactivity, emesis, and salivation were observed in dogs; and changes in fecal consistency were noted in all species except mice. Increased kidney weights in male rats correlated with the accumulation of hyaline droplets in renal proximal tubular epithelium. No changes were found in the kidneys of female rats. Reversible increases in liver weight were observed in rats administered gabapentin at 3,000 mg/kg for 13 weeks or 1,500 mg/kg for 26 weeks; and in dogs at 2,000 mg/kg for 6 months. No pathologic findings were noted in mice given up to 2,000 mg/kg gabapentin for 13 weeks or in monkeys given up to 500 mg/kg for 52 weeks.
In rats, plasma gabapentin concentrations increased with increasing dose. The increases were not dose proportional between 2,000 and 3,000 mg/kg, suggesting saturation of absorption at high doses.
Carcinogenesis and Mutagenesis: Gabapentin was given in the diet to mice at 200, 600, and 2,000 mg/kg/day and to rats at 250, 1,000, and 2,000 mg/kg/day for 2 years. A statistically significant increase in the incidence of pancreatic acinar cell tumours was found only in male rats at the highest dose, but not in female rats or in mice of either sex. Peak plasma drug concentrations and areas under the concentration time curve in rats at 2,000 mg/kg are 20 times higher than the therapeutic concentrations in humans given 1,200 mg/day and are 14 times higher than the therapeutic concentrations in humans given 2,400 mg/day.
The pancreatic acinar cell tumours in male rats are low grade malignancies, did not affect survival, did not metastasize or invade surrounding tissue, and were similar to those seen in concurrent controls. Furthermore, higher concentrations of gabapentin in pancreas relative to plasma have been observed in rats but not monkeys, which may account for the species-specific effects.
The relevance of these pancreatic acinar cell tumours in male rats to carcinogenic risk in humans is unclear, as the biologic characteristics of the tumours in rats are unlike those observed in humans. Ductal carcinoma comprises over 90% of all primary cancers of human exocrine pancreas, whereas acinar cell adenomas represent the primary pancreatic exocrine tumours in rats. In humans, pancreatic neoplasia exhibits local and distant tumour spread at the time of diagnosis. Metastasis occurs in 67% of cases, and survival is between 2 and 6 months after diagnosis. In contrast, pancreatic acinar cell tumours in male rats given gabapentin did not metastasize, exhibit aggressive behaviour or affect survival.
Gabapentin has no genotoxic potential. It was not mutagenic in the Ames bacterial plate incorporation assay or at the HGPRT locus in mammalian cells in the presence or absence of metabolic activation. Gabapentin did not induce structural chromosome aberrations in mammalian cells in vitro or in vivo, and did not induce micronucleus formation in the bone marrow of hamsters.
Reproduction Studies: In a fertility and general reproduction study in rats with dietary doses of gabapentin up to 2,000 mg/kg, (approximately 5 times the maximum daily human dose, on a mg/m2 basis), no adverse effects were noted on fertility, precoital interval, pregnancy rate, gestation length, parturition, nesting/nursing behaviour, or lactation.
Gabapentin did not increase the incidence of malformations, compared to controls, in the offsprings of mice, rats, or rabbits at doses up to 50, 30, and 25 times, respectively, the daily human dose of 3,600 mg, (4, 5 or 8 times, respectively, the human daily dose, on a mg/m2 basis).
When pregnant mice received oral doses of gabapentin (500, 1,000 or 3,000 mg/kg/day) during the period of organogenesis embryofetal toxicity (increased incidence of skeletal variations) was observed at 1,000 and 3,000 mg/kg/day (17 and 50 times, respectively the human daily dose of 3,600 mg; 1.3 and 4 times, respectively, the human daily dose on a mg/m2 basis). The no-effect dose for embryofetal developmental toxicity in mice was observed at 500 mg/kg/day (8 times the human daily dose of 3,600 mg; 0.7 times the human daily dose, on a mg/m2) basis.
In studies in which rats received oral doses of gabapentin (500 to 2,000 mg/kg/day) during pregnancy, adverse effect on offspring development (increased incidence of hydroureter and/or hydronephrosis) were observed at all doses. The lowest dose tested is similar to the MRHD on a mg/m2 basis.
When pregnant rabbits were treated with gabapentin during the period of organogenesis, an increase in embryofetal mortality was observed at all doses tested (60, 300, or 1,500 mg/kg). The lowest dose tested is less than the MRHD on a mg/m2 basis.
In a published study, gabapentin (400 mg/kg/day) was administered by intraperitoneal injection to neonatal mice during the first postnatal week; a period of synaptogenesis in rodents (corresponding to the last trimester of pregnancy in humans). Gabapentin caused a marked decrease in neuronal synapse formation in brains of intact mice and abnormal neuronal synapse formation in a mouse model of synaptic repair. Gabapentin has been shown in vitro to interfere with activity of the α2δ subunit of voltage-activated calcium channels; a receptor involved in neuronal synaptogenesis. The clinical significance of these findings is unknown.