


 Sign Out
Sign Out


Pharmacology: Pharmacodynamics: Lenvatinib is a multikinase inhibitor which has shown mainly antiangiogenic properties in vitro and in vivo, and direct inhibition of tumour growth was also observed in in vitro models.
Mechanism of action: Lenvatinib is a receptor tyrosine kinase (RTK) inhibitor that selectively inhibits the kinase activities of vascular endothelial growth factor (VEGF) receptors VEGFR1 (FLT1), VEGFR2 (KDR), and VEGFR3 (FLT4), in addition to other proangiogenic and oncogenic pathway-related RTKs including fibroblast growth factor (FGF) receptors FGFR1, 2, 3, and 4, the platelet derived growth factor (PDGF) receptor PDGFRα, KIT, and RET. Lenvatinib also exhibited antiproliferative activity in hepatocellular carcinoma cell lines dependent on activated FGFR signaling with a concurrent inhibition of FGF-receptor substrate 2α (FRS2α) phosphorylation.
In syngeneic mouse tumor models, lenvatinib decreased tumor-associated macrophages, increased activated cytotoxic T cells, and demonstrated greater antitumor activity in combination with an antiPD-1 monoclonal antibody compared to either treatment alone.
The combination of lenvatinib and everolimus showed increased antiangiogenic and antitumour activity as demonstrated by decreases in human endothelial cell proliferation, tube formation, and VEGF signaling in vitro, and by decreases in tumour volume in mouse xenograft models of human renal cell cancer that were greater than those with either drug alone.
Although not studied directly with lenvatinib, the mechanism of action (MOA) for hypertension is postulated to be mediated by the inhibition of VEGFR2 in vascular endothelial cells. Similarly, although not studied directly, the MOA for proteinuria is postulated to be mediated by downregulation of VEGFR1 and VEGFR2 in the podocytes of the glomerulus.
The mechanism of action for hypothyroidism is not fully elucidated.
Clinical efficacy: The clinical safety and efficacy of LENVIMA have been studied in patients with differentiated thyroid cancer, renal cell carcinoma and hepatocellular carcinoma.
Radioactive iodine-refractory differentiated thyroid cancer: The SELECT study was a multicentre, randomised, double-blind, placebo-controlled trial that was conducted in 392 patients with radioactive iodine-refractory differentiated thyroid cancer with independent, centrally reviewed, radiographic evidence of disease progression within 12 months (+1month window) prior to enrollment. Radioactive iodine-refractory was defined as one or more measurable lesions either with a lack of iodine uptake or with progression in spite of radioactive-iodine (RAI) therapy, or having a cumulative activity of RAI of >600 mCi or 22 GBq with the last dose at least 6 months prior to study entry. Randomisation was stratified by geographic region (Europe, North America, and Other), prior VEGF/VEGFR-targeted therapy (patients may have received 0 or 1 prior VEGF/VEGFR-targeted therapy), and age (≤65 years or >65 years). The main efficacy outcome measure was progression-free survival (PFS) as determined by blinded independent radiologic review using Response Evaluation Criteria in Solid Tumours (RECIST) 1.1. Secondary efficacy outcome measures included overall response rate and overall survival. Patients in the placebo arm could opt to receive lenvatinib treatment at the time of confirmed disease progression.
Eligible patients with measurable disease according to RECIST 1.1 were randomised 2:1 to receive lenvatinib 24 mg once daily (n=261) or placebo (n=131). Baseline demographics and disease characteristics were well balanced for both treatment groups. Of the 392 patients randomised, 76.3% were naïve to prior VEGF/VEGFR-targeted therapies, 49.0% were female, 49.7% were European, and the median age was 63 years. Histologically, 66.1% had a confirmed diagnosis of papillary thyroid cancer and 33.9% had follicular thyroid cancer which included Hürthle cell 14.8% and clear cell 3.8%. Metastases were present in 99% of the patients: lungs in 89.3%, lymph nodes in 51.5%, bone in 38.8%, liver in 18.1%, pleura in 16.3%, and brain in 4.1%. The majority of patients had an ECOG performance status of 0; 42.1% had a status of 1; 3.9% had a status above 1. The median cumulative RAI activity administered prior to study entry was 350 mCi (12.95 GBq).
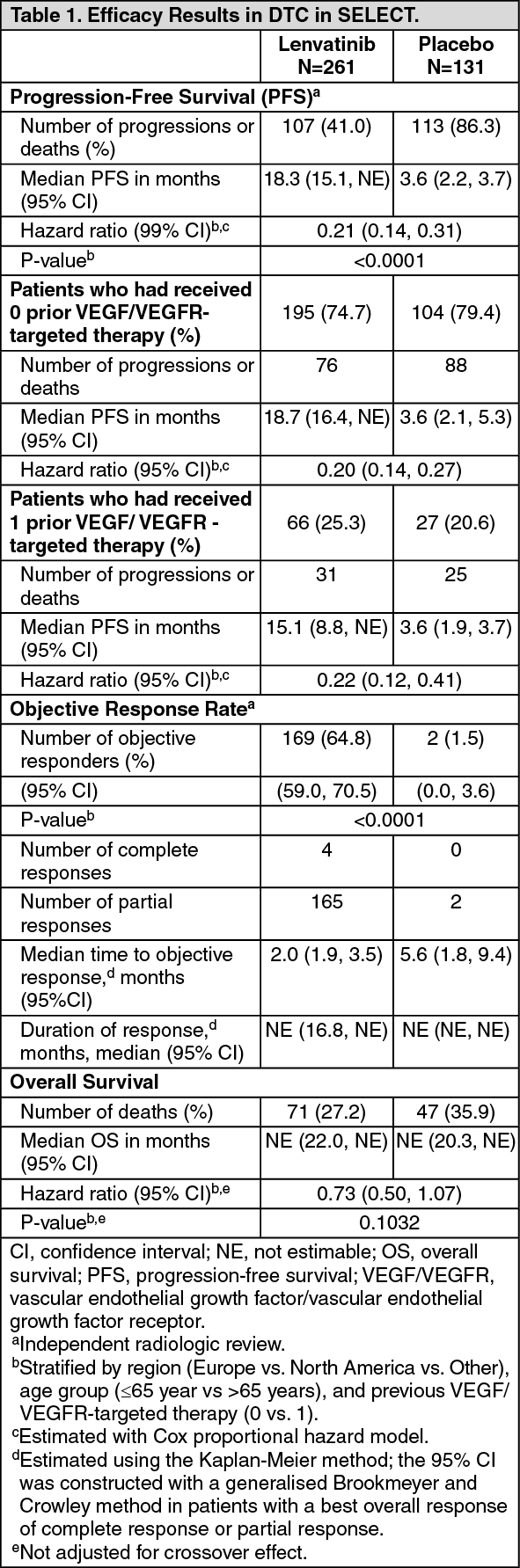
A statistically significant prolongation in PFS was demonstrated in lenvatinib-treated patients compared with those receiving placebo (p<0.0001) (see Figure 1). The positive effect on PFS was seen across the subgroups of age (above or below 65 years), sex, race, histological subtype, geographic region, and those who received 0 or 1 prior VEGF/VEGFR-targeted therapy (see Table 1). Following independent review confirmation of disease progression, 109 (83.2%) patients randomised to placebo crossed over to open-label lenvatinib at the time of the primary efficacy analysis.
The objective response rate (complete response [CR] plus partial response [PR]) per independent radiological review was significantly (p<0.0001) higher in the lenvatinib-treated group (64.8%) than in the placebo-treated group (1.5%). Four (1.5%) subjects treated with lenvatinib attained a CR and 165 subjects (63.2%) had a PR, while no subjects treated with placebo had a CR and 2 (1.5%) subjects had a PR.
The median time to first dose reduction was 2.8 months. The median time to objective response was 2.0 (95% CI: 1.9, 3.5) months; however, of the patients who experienced a complete or partial response to lenvatinib, 70.4% were observed to develop the response on or within 30 days of being on the 24-mg dose.
An overall survival analysis was confounded by the fact that placebo-treated subjects with confirmed disease progression had the option to cross over to open-label lenvatinib. There was no statistically significant difference in overall survival between the treatment groups at the time of the primary efficacy analysis (HR=0.73; 95% CI: 0.50, 1.07, p=0.1032). The median OS had not been reached for either the lenvatinib group or the placebo crossover group. (See Table 1 and Figure 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image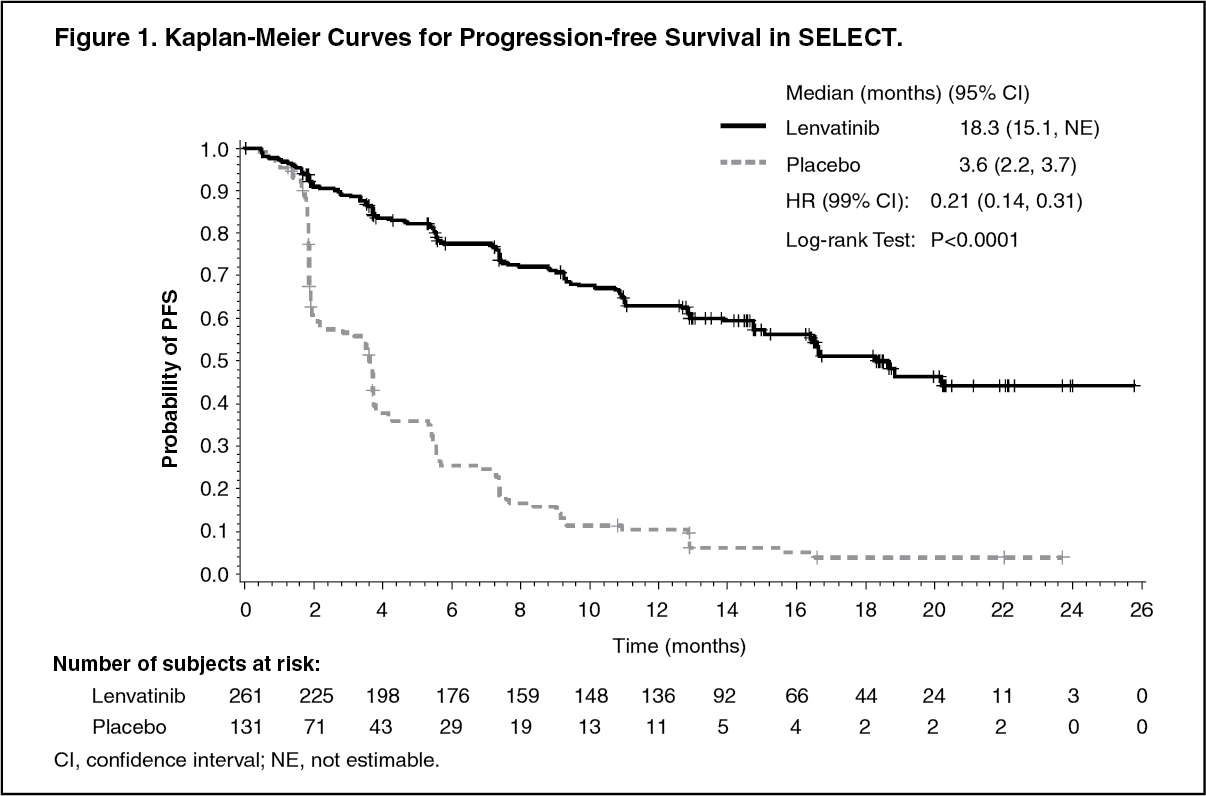 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageRenal cell carcinoma: In combination with pembrolizumab: First-line treatment (Study 307): The efficacy of lenvatinib in combination with pembrolizumab was investigated in (CLEAR, Study 307), a multicenter, open-label, randomized trial that enrolled 1069 patients with advanced RCC in the first-line setting. Patients were enrolled regardless of PD-L1 tumor expression status. Patients with active autoimmune disease or a medical condition that required immunosuppression were ineligible. Randomization was stratified by geographic region. (North America and Western Europe versus "Rest of the World") and Memorial Sloan Kettering Cancer Center (MSKCC) prognostic groups (favorable, intermediate, and poor risk).
Patients were randomized to lenvatinib 20 mg orally once daily in combination with pembrolizumab 200 mg intravenously every 3 weeks (n=355), or lenvatinib 18 mg orally once daily in combination with everolimus 5 mg orally once daily (n=357), or sunitinib 50 mg orally once daily for 4 weeks then off treatment for 2 weeks (n=357). All patients on the lenvatinib plus pembrolizumab arm were started on lenvatinib 20 mg orally once daily. The median time to first dose reduction for lenvatinib was 1.9 months. The median average daily dose for lenvatinib was 14 mg. Treatment continued until unacceptable toxicity or disease progression as determined by the investigator and confirmed by independent radiologic review committee (IRC) using Response Evaluation Criteria in Solid Tumors Version 1.1 (RECIST 1.1).
Administration of lenvatinib with pembrolizumab was permitted beyond RECIST-defined disease progression if the patient was clinically stable and considered by the investigator to be deriving clinical benefit. Pembrolizumab dosing was continued for a maximum of 24 months; however, treatment with lenvatinib could be continued beyond 24 months. Assessment of tumor status was performed at baseline and then every 8 weeks.
The overall study population characteristics were: median age of 62 years (range: 29 to 88 years); 42% age 65 or older, 75% male; 74% White, 21% Asian, 1% Black, and 2% other races; 18% and 82% of patients had a baseline KPS of 70 to 80 and 90 to 100, respectively; patient distribution by IMDC (International Metastatic RCC Database Consortium) risk categories was 33% favorable, 56% intermediate and 10% poor, and MSKCC risk categories was 27% favorable, 64% intermediate and 9% poor. Common sites of metastases in patients were lung (68%), lymph node (45%), and bone (25%).
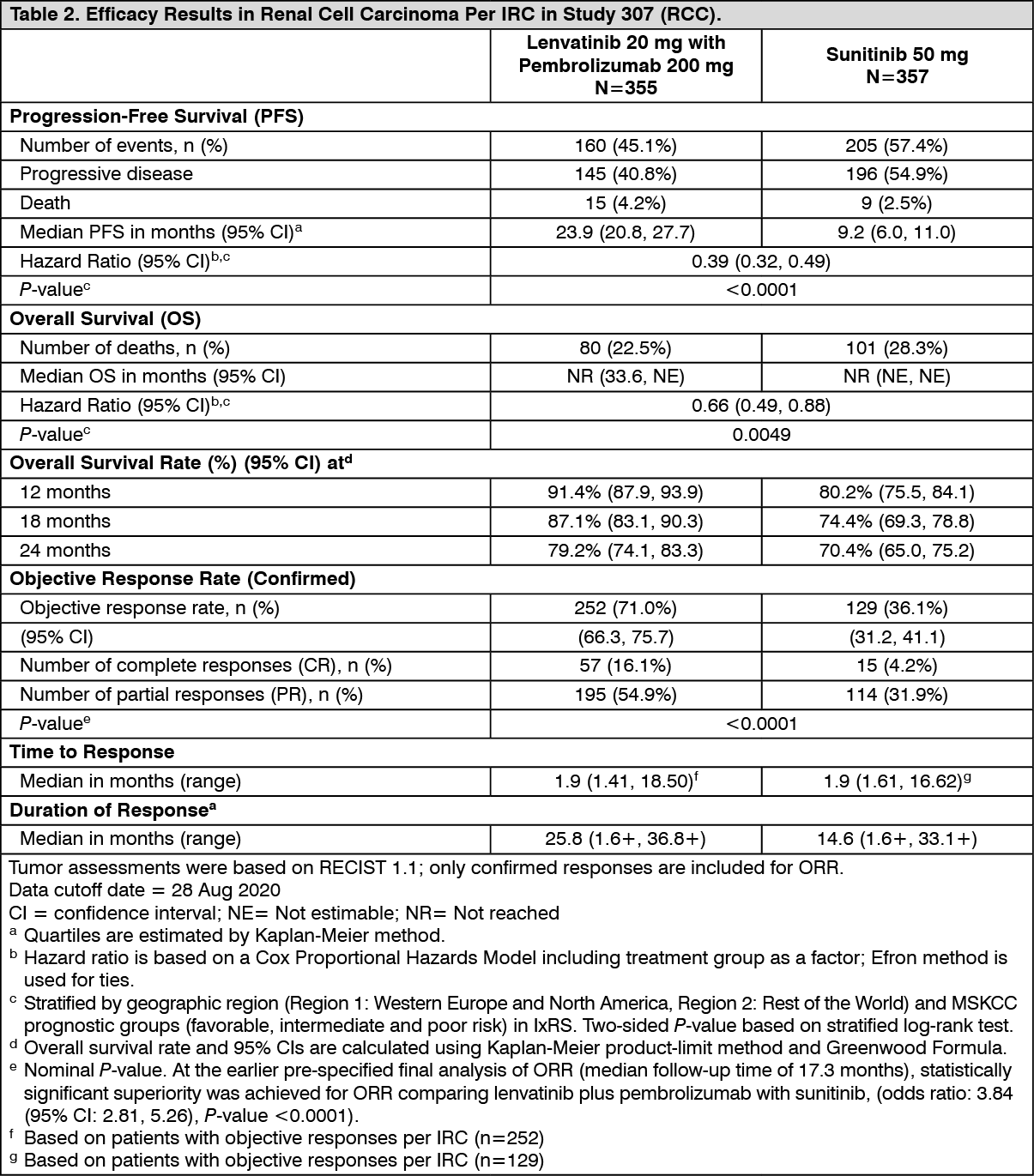
The primary efficacy outcome measure was progression free survival (PFS) based on RECIST 1.1 per IRC. Key secondary efficacy outcome measures included overall survival (OS) and objective response rate (ORR). Lenvatinib in combination with pembrolizumab demonstrated statistically significant improvements in PFS, OS and ORR compared with sunitinib. Efficacy results for CLEAR are summarized in Table 2 and Figures 2, 3 and 4, at a median OS follow-up time of 26.6 months. Progressive disease as best overall response was observed in 5.4% of patients treated with lenvatinib in combination with pembrolizumab compared with 14.0% of patients treated with sunitinib. Consistent results were observed across pre-specified subgroups, MSKCC prognostic groups and PD-L1 tumor expression status (see Figure 3). (See Table 2 and Figures 2, 3 and 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image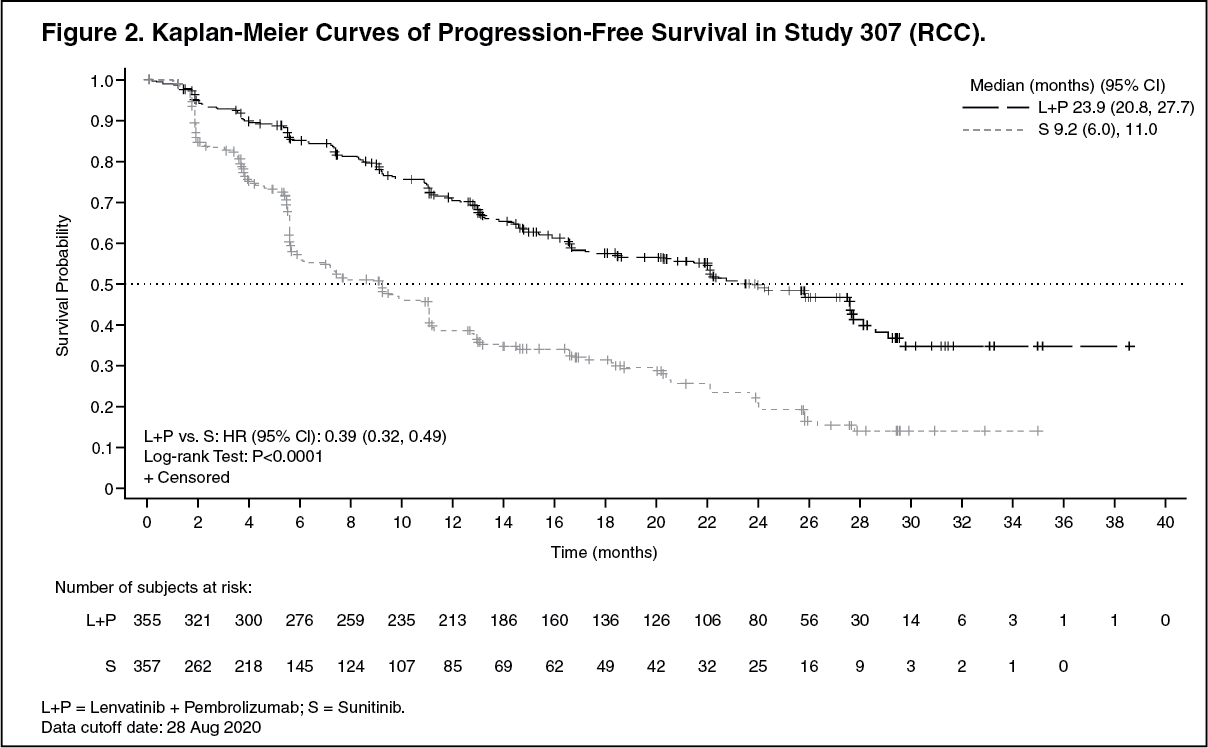 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image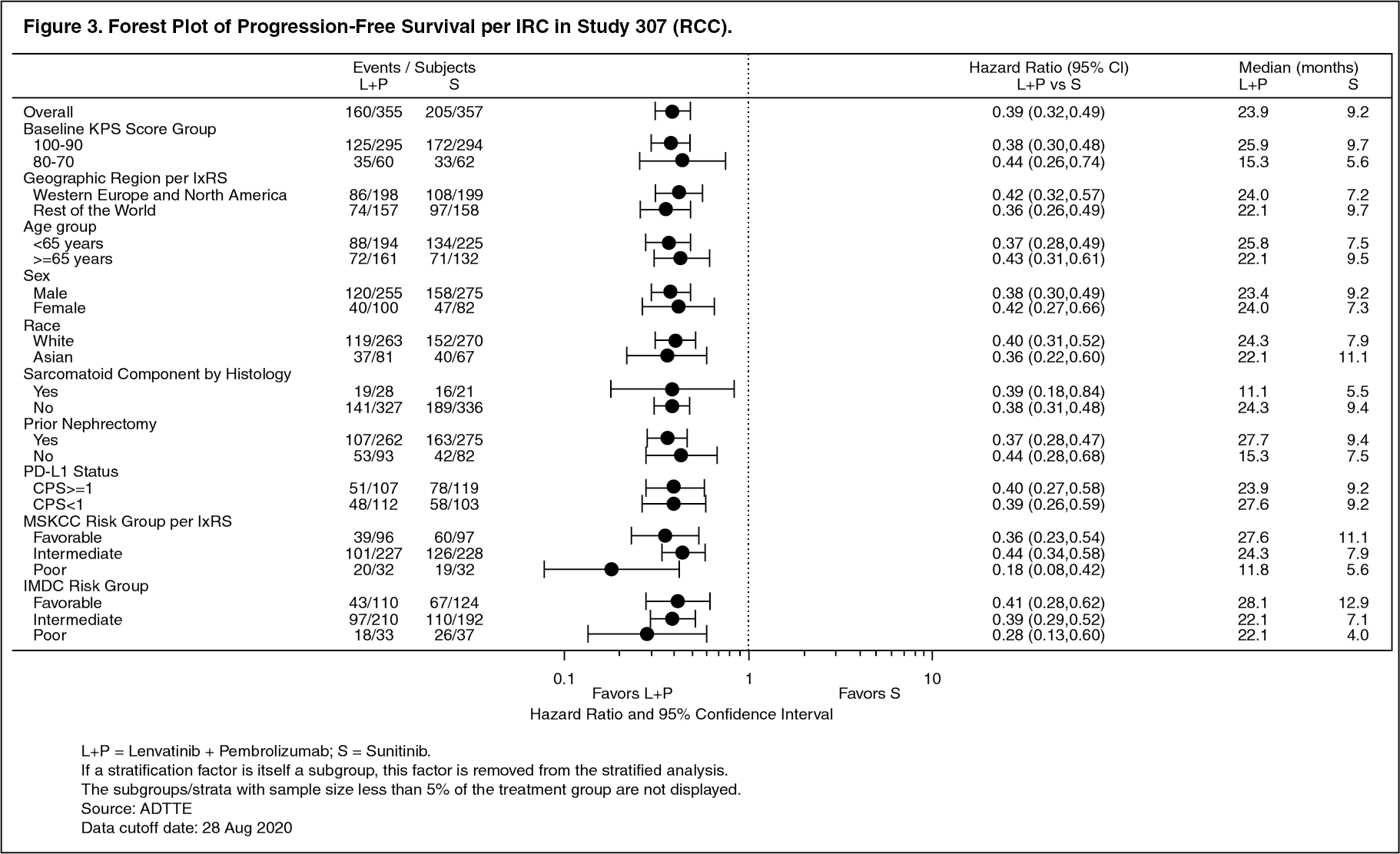 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image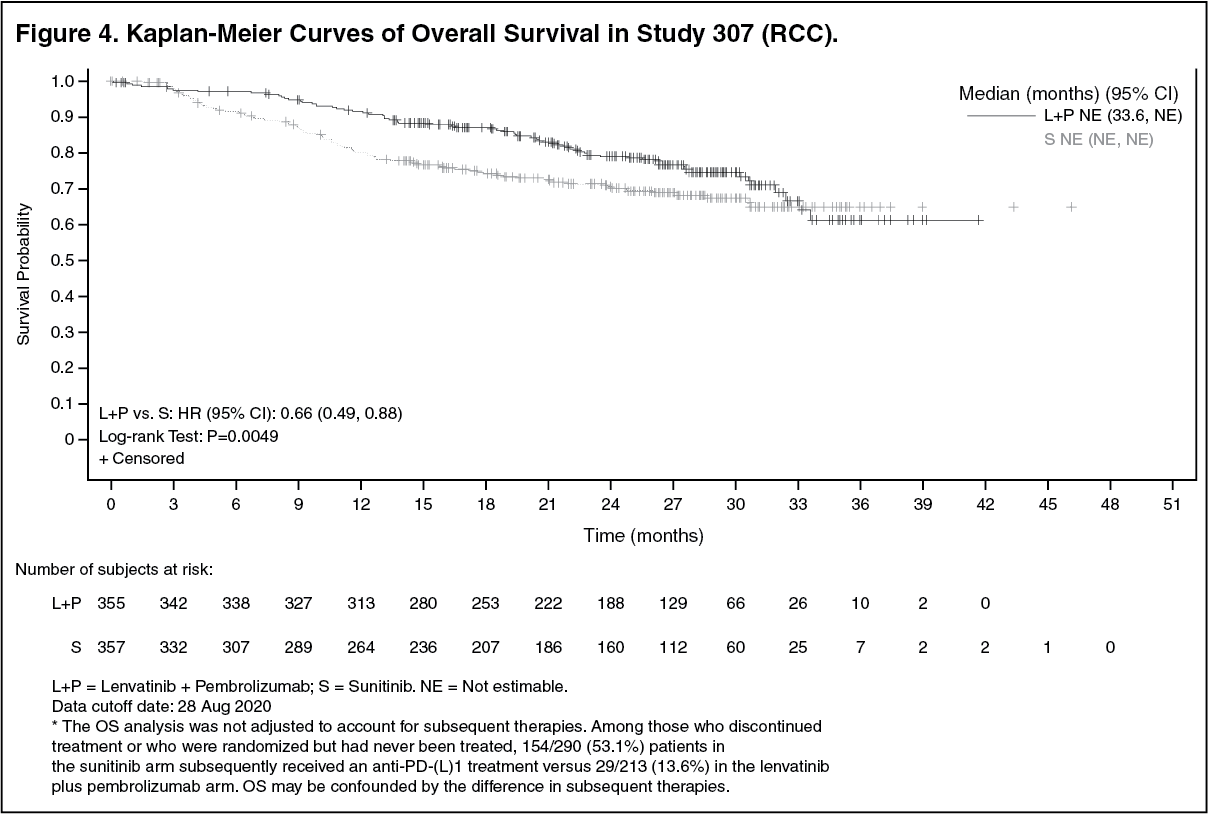 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAssessment of Quality of Life (QoL) in patients with RCC: Patient-reported outcomes (PRO) were assessed using the European Organization for the Research and Treatment of Cancer (EORTC) QLQ-30 and Kidney Cancer Symptom Index (FKSI-DRS). From baseline to a mean follow-up time of 46 weeks, patients treated with lenvatinib in combination with pembrolizumab had better physical functioning, fatigue, dyspnea, and constipation scores compared to the sunitinib group.
Compared to sunitinib, lenvatinib in combination with pembrolizumab showed a more than 12 week delay in median time to worsening in global health status (GHS), physical functioning and patient reported symptoms with no subsequent recovery: EORTC QLQ-C30 GHS (114 vs. 75 weeks, HR=0.6 [95%CI: 0.47, 0.77]), physical functioning (134 vs 78 weeks, HR=0.52 [95% CI: 0.41, 0.67]), fatigue (110 vs. 59 weeks, HR=0.54 [95% CI: 0.43, 0.67]), insomnia (156 vs. 126 weeks, HR=0.63 [95% CI: 0.47, 0.85]), dyspnea (153 vs. 126 weeks, HR=0.56 [95% CI: 0.41, 0.76]), nausea and vomiting (147 vs. 131 weeks, (HR=0.53 [95% CI: 0.39, 0.74]), pain (119 vs. 105 weeks, HR=0.68 [95% CI: 0.53, 0.87]) and FKSI-DRS (134 vs. 117 weeks, HR=0.7 [95% CI: 0.53, 0.92]).
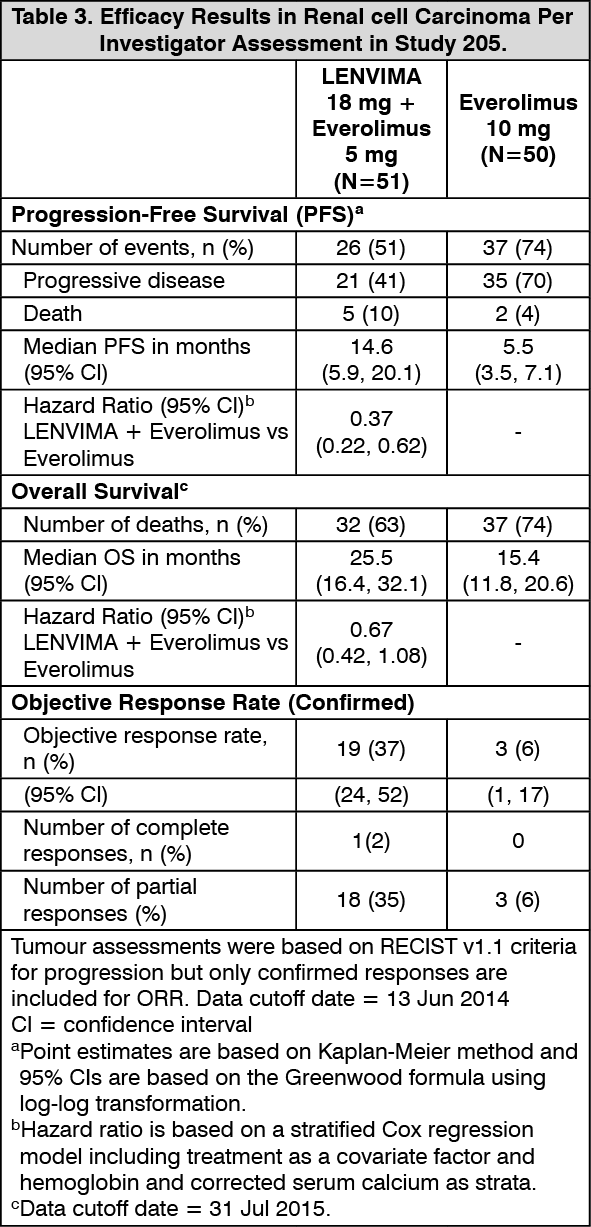
In combination with everolimus: Second-line treatment (Study 205): A multicenter study (Study 205) randomized 153 patients with advanced or metastatic renal cell carcinoma who have previously received anti-angiogenic therapy 1:1:1 to LENVIMA 18 mg plus everolimus 5 mg, LENVIMA 24 mg monotherapy, or everolimus 10 mg monotherapy. All medications were administered orally once daily. Patients were required to have histological confirmation of clear cell RCC and ECOG Performance Status of 0 or 1. Patients were stratified by hemoglobin level (≤ or > 13 g/dL for males and ≤ or > 11.5 g/dL for females) and corrected serum calcium (≥ 10 mg/dL vs. < 10 mg/dL).
Of the 101 patients randomly allocated to the LENVIMA + everolimus arm and everolimus monotherapy arm, 72% were male, the median age was 60 years, 31% were older than 65 years, 96% were White. Metastases were present in 95% of the patients and unresectable advanced disease was present in 5%. All patients had a baseline ECOG PS of either 0 (54%) or 1 (46%) with similar distribution across the 2 treatment arms. Memorial Sloan Kettering Cancer Center (MSKCC) favorable, intermediate, and poor risk categories were observed respectively, in 24%, 37%, and 39% of patients in the LENVIMA + everolimus arm, and 24%, 38%, and 38% of patients in the everolimus arm.
The major efficacy outcome measure was investigator-assessed PFS evaluated according to RECIST 1.1. Efficacy results from Study 2 are summarized in Table 3 and Figures 5 and 6. The treatment effect of the combination on PFS was supported by a retrospective independent review of radiographs with an observed hazard ratio (HR) of 0.43 (95% CI: 0.24, 0.75) compared with the everolimus arm. (See Table 3 and Figures 5 and 6.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image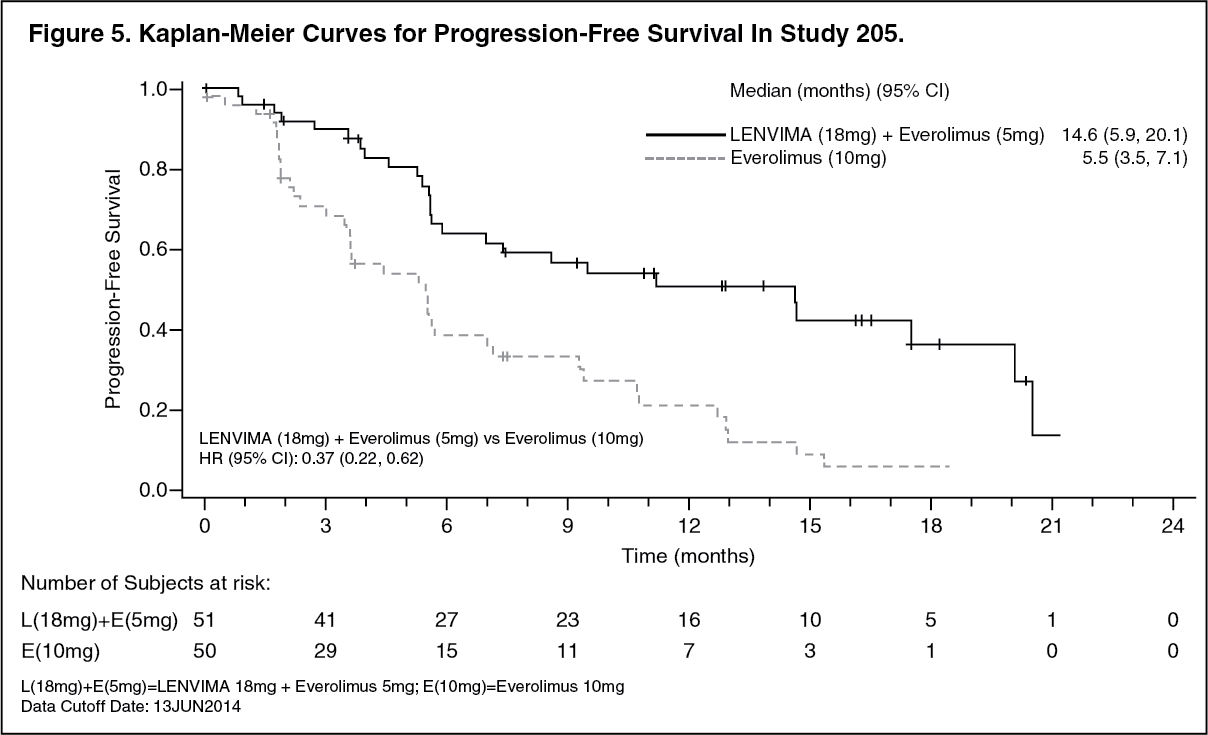 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image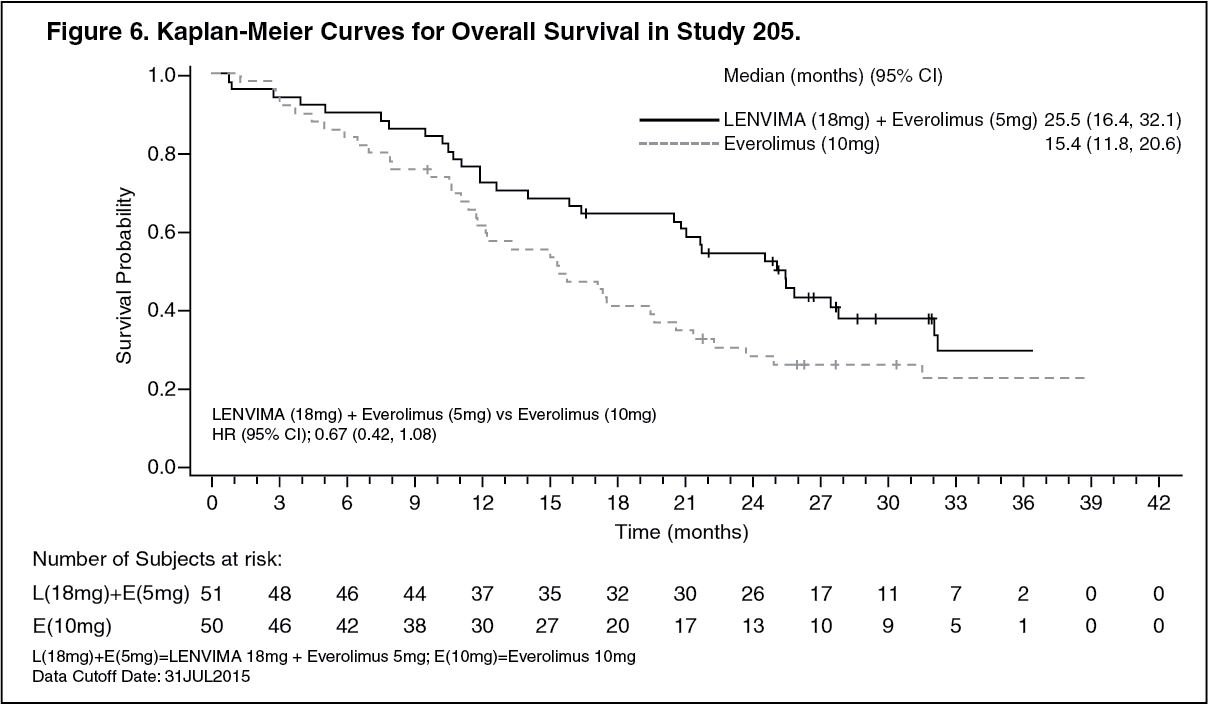 Click on icon to see table/diagram/image
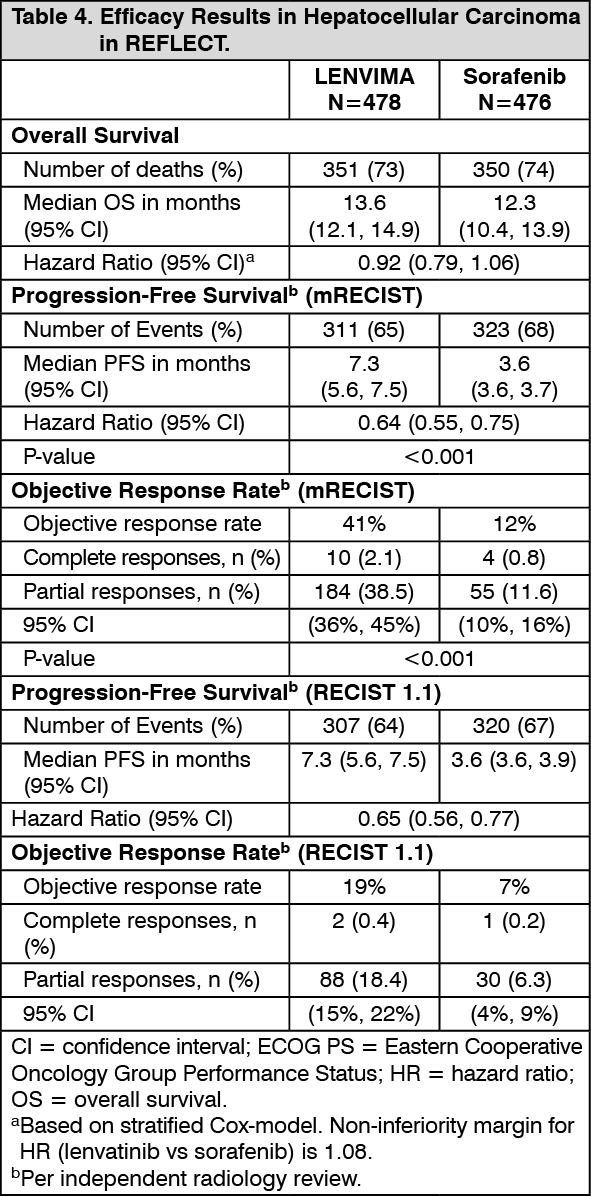
Click on icon to see table/diagram/imageHepatocellular carcinoma: The efficacy of LENVIMA was evaluated in a randomized, open-label, multicenter, international study (REFLECT; NCT01761266) conducted in patients with previously untreated unresectable hepatocellular carcinoma (HCC). The study enrolled adults with Child-Pugh A and Barcelona Clinic Liver Cancer (BCLC) Stage C or B HCC who were ineligible for local liver-directed therapy; had an ECOG PS of 0 or 1; had received no prior systemic therapy for HCC; and had at least one measurable target lesion according to modified RECIST for HCC.
Patients were randomized (1:1) to receive LENVIMA (12 mg for baseline body weight ≥60 kg or 8 mg for baseline body weight <60 kg) orally once daily or sorafenib 400 mg orally twice daily until radiological disease progression or unacceptable toxicity. Randomization was stratified by region (Western vs Asia Pacific), presence of macroscopic portal vein invasion or extrahepatic spread (yes vs no), ECOG PS (0 vs 1), and body weight (<60 kg vs ≥60 kg). The major efficacy outcome measure was overall survival (OS). REFLECT was designed to show the non-inferiority of LENVIMA to sorafenib for OS. Additional efficacy outcome measures were progression-free survival (PFS) and objective response rate (ORR) according to modified RECIST for HCC.
A total of 954 patients were randomized, 478 to the LENVIMA arm and 476 to the sorafenib arm. The demographics of the study population were: median age of 62 years (range: 20 to 88 years); 84% male; 69% Asian and 29% White; 63% ECOG PS of 0; and 69% weighed ≥60 kg. Of the 590 (62%) patients with at least one site of documented distant metastatic disease, 52% had lung metastasis, 45% had lymph node metastasis, and 16% had bone metastasis.
Macroscopic portal vein invasion, extra-hepatic spread, or both were present in 70% of patients. HCC was categorized as Child-Pugh A and BCLC Stage C in 79% and Child-Pugh A and BCLC Stage B in 21% of patients. Seventy-five percent (75%) of patients had radiographic evidence of cirrhosis at baseline. Investigator-documented primary risk factors for the development of HCC were hepatitis B (50%), hepatitis C (23%), alcohol use (6%), other (7%), and unknown (14%).
REFLECT demonstrated that LENVIMA was non-inferior to sorafenib for OS. REFLECT did not demonstrate a statistically significant improvement in OS for patients randomized to LENVIMA as compared to those in the sorafenib arm. LENVIMA was statistically significantly superior to sorafenib for PFS and ORR. Efficacy results are summarized in Table 4 and Figure 7. (See Table 4 and Figure 7.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image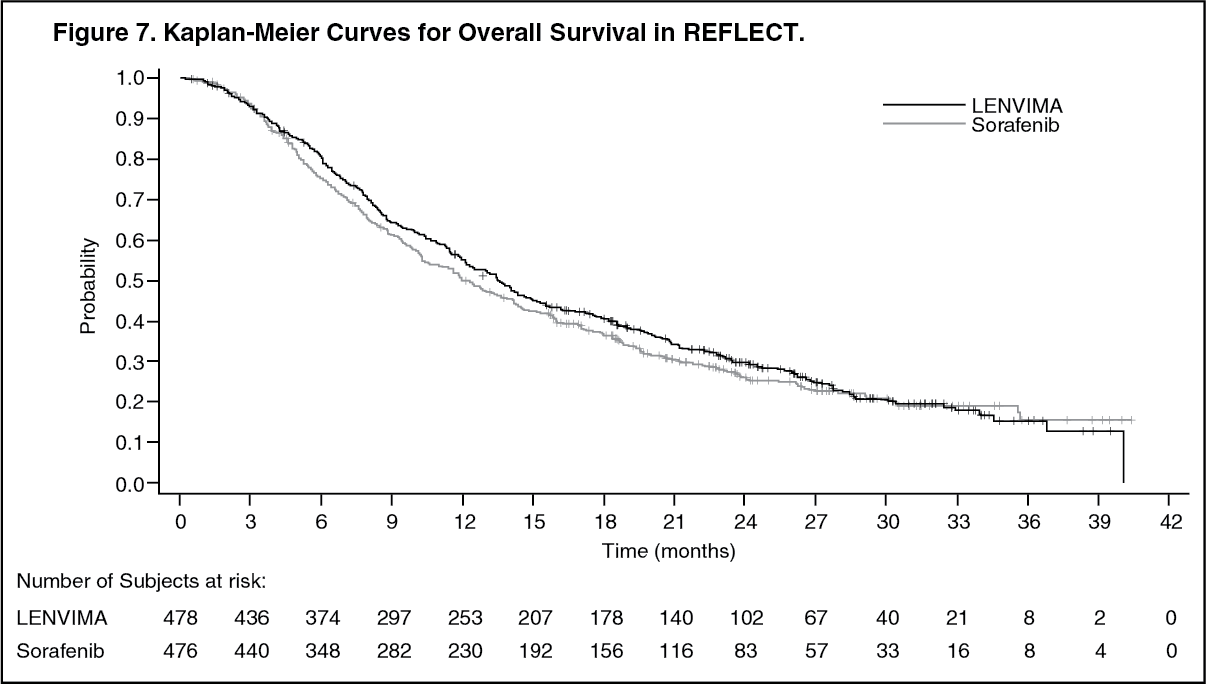 Click on icon to see table/diagram/image
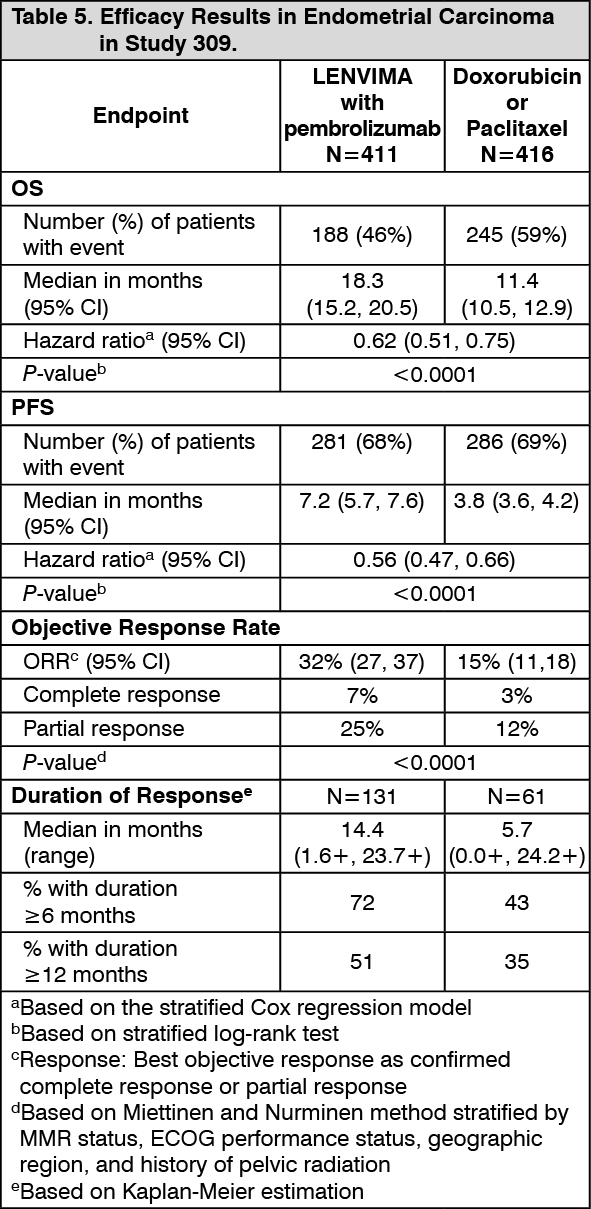
Click on icon to see table/diagram/imageEndometrial Carcinoma (EC): The efficacy of lenvatinib in combination with pembrolizumab was investigated in Study 309, a randomized, multicenter, open-label, active-controlled study conducted in patients with advanced EC who had been previously treated with at least one prior platinum-based chemotherapy regimen in any setting, including in the neoadjuvant and adjuvant settings. The study excluded patients with endometrial sarcoma (including carcinosarcoma), or patients who had active autoimmune disease or a medical condition that required immunosuppression. Randomization was stratified by MMR status (dMMR or pMMR [not dMMR]) using an IHC test. The pMMR stratum was further stratified by ECOG performance status, geographic region, and history of pelvic radiation. Patients were randomised (1:1) to one of the following treatment arms: lenvatinib 20 mg orally once daily in combination with pembrolizumab 200 mg intravenously every 3 weeks; investigator's choice consisting of either doxorubicin 60 mg/m2 every 3 weeks, or paclitaxel 80 mg/m2 given weekly, 3 weeks on/1 week off.
Treatment with lenvatinib and pembrolizumab continued until RECIST v1.1-defined progression of disease as verified by blinded independent central review (BICR), unacceptable toxicity, or for pembrolizumab, a maximum of 24 months. Administration of study treatment was permitted beyond RECIST-defined disease progression if the treating investigator considered the patient to be deriving clinical benefit and the treatment was tolerated. Assessment of tumor status was performed every 8 weeks.
A total of 827 patients were enrolled and randomized to lenvatinib in combination with pembrolizumab (n=411) or investigator's choice of doxorubicin (n=306) or paclitaxel (n=110). The baseline characteristics of these patients were: median age of 65 years (range 30 to 86), 50% age 65 or older; 61% White, 21% Asian, and 4% Black; ECOG PS of 0 (59%) or 1 (41%), and 84% with pMMR tumor status. The histologic subtypes were endometrioid carcinoma (60%), serous (26%), clear cell carcinoma (6%), mixed (5%), and other (3%). All 827 of these patients received prior systemic therapy for EC: 69% had one, 28% had two, and 3% had three or more prior systemic therapies. Thirty-seven percent of patients received only prior neoadjuvant or adjuvant therapy.
The primary efficacy outcome measures were OS and PFS (as assessed by BICR using RECIST 1.1). Secondary efficacy outcome measures included ORR, as assessed by BICR using RECIST 1.1. The median follow-up time was 11.4 months (range: 0.3 to 26.9 months). Efficacy results for Study 309 are summarized in Table 5 and Figures 8, 9, and 10. (See Table 5 and Figures 8, 9, and 10.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image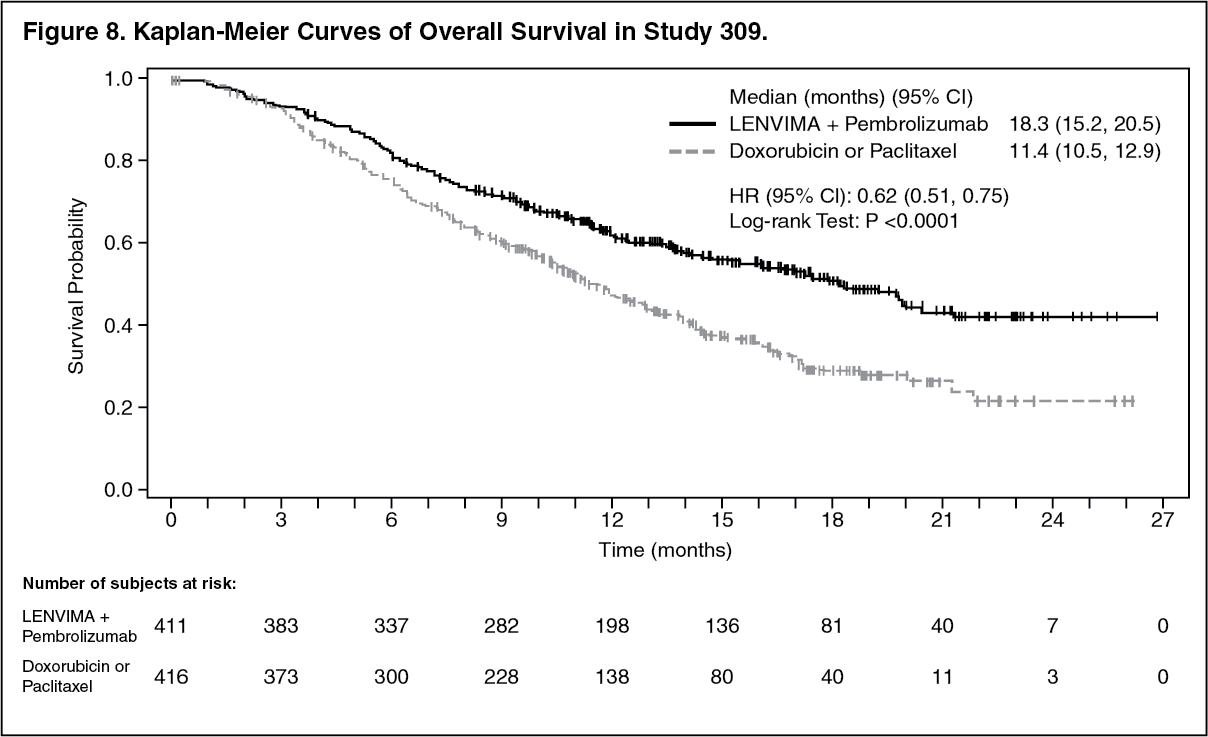 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image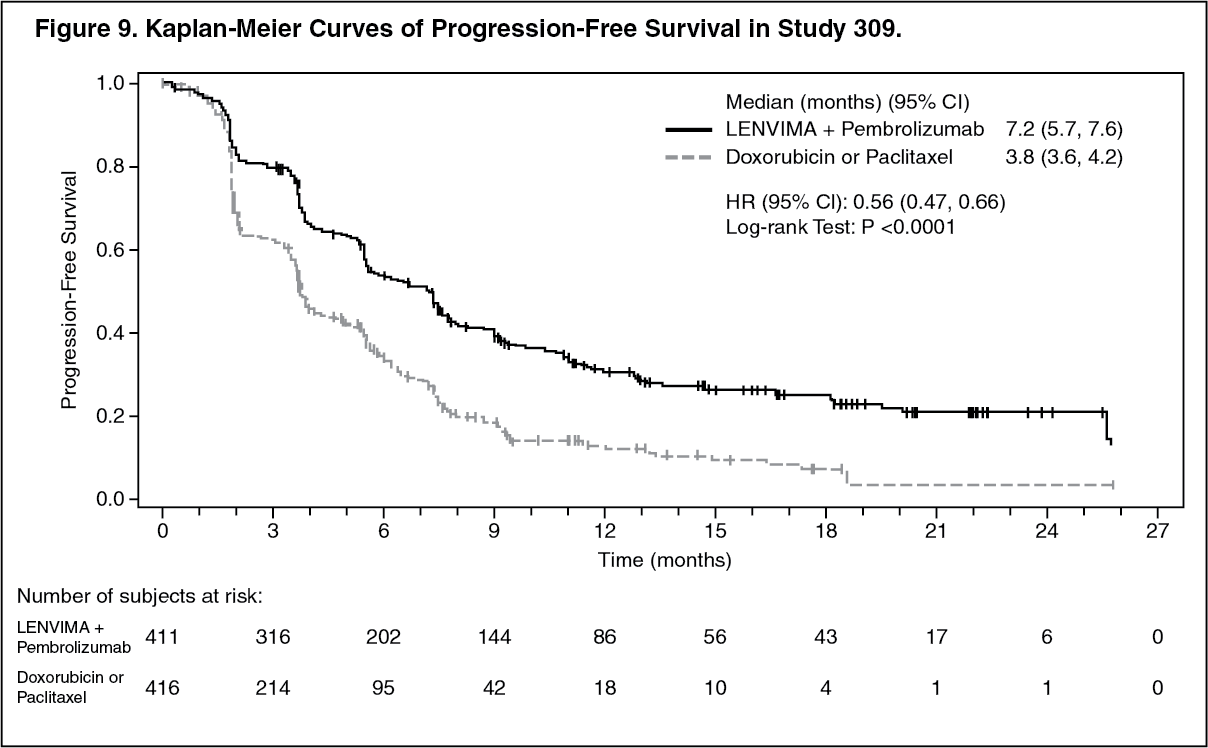 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image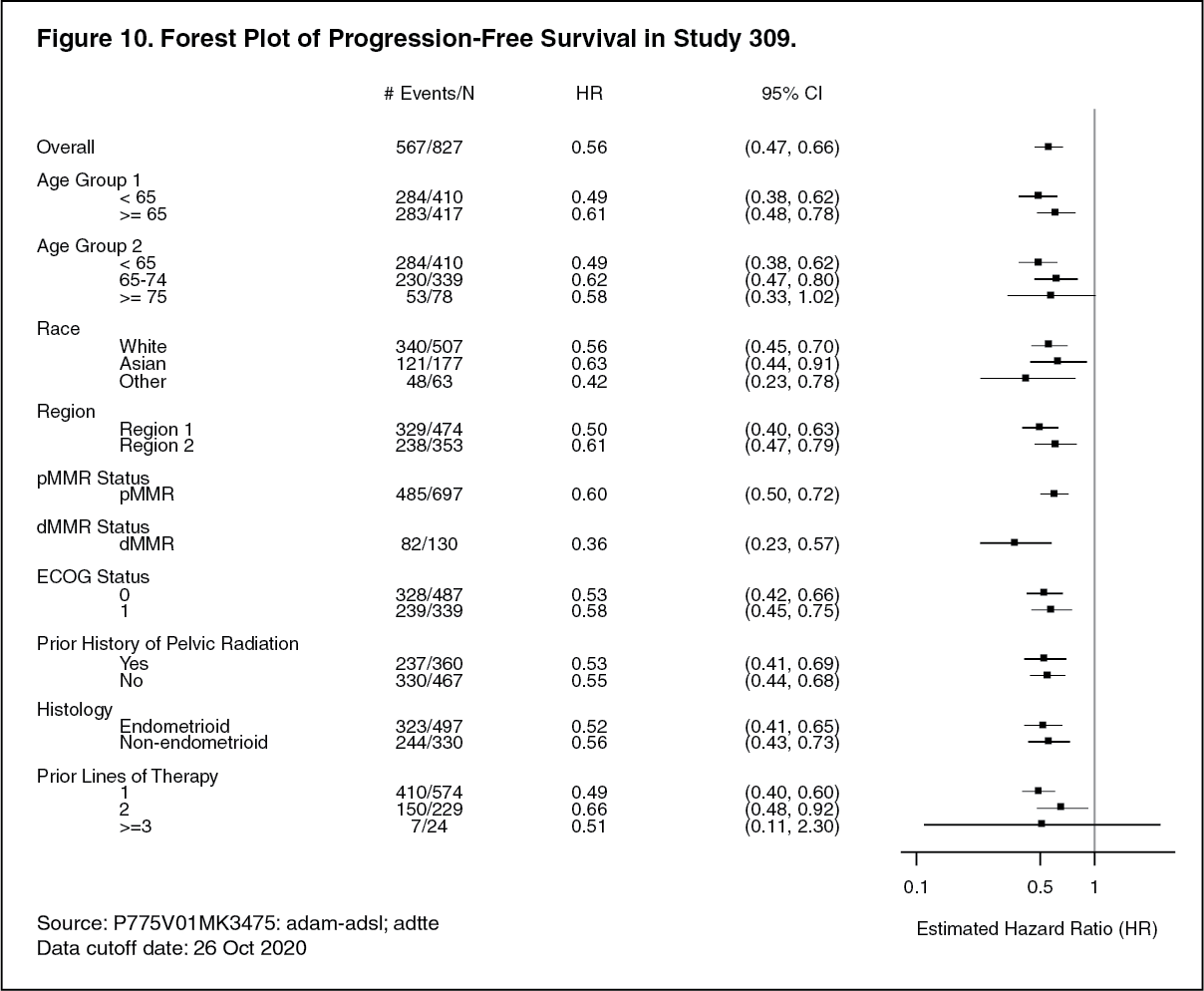 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePharmacokinetics: Pharmacokinetic parameters of lenvatinib have been studied in healthy adult subjects, adult subjects with hepatic impairment, renal impairment, and solid tumours.
Absorption: Lenvatinib is rapidly absorbed after oral administration with tmax typically observed from 1 to 4 hours postdose. Food does not affect the extent of absorption, but slows the rate of absorption. When administered with food to healthy subjects, peak plasma concentrations are delayed by 2 hours. Absolute bioavailability has not been determined in humans; however, data from a mass-balance study suggests that it is in the order of 85%. Lenvatinib exhibited good oral bioavailability in dogs (70.4%) and monkeys (78.4%).
Distribution: In vitro binding of lenvatinib to human plasma proteins is high and ranged from 98% to 99% (0.3 - 30 μg/mL, mesilate). This binding was mainly to albumin with minor binding to α1-acid glycoprotein and γ-globulin. A similar plasma protein binding (97% to 99%) with no dependencies on lenvatinib concentrations (0.2 to 1.2 μg/mL) was observed in plasma from hepatically impaired, renally impaired, and matching healthy subjects.
In vitro, the lenvatinib blood-to-plasma concentration ratio ranged from 0.589 to 0.608 (0.1 - 10 μg/mL, mesilate).
Lenvatinib is a substrate for P-gp and BCRP. Lenvatinib is not a substrate for OAT1, OAT3, OATP1B1, OATP1B3, OCT1, OCT2, MATE1, MATE2-K or the bile salt export pump BSEP.
In patients, the median apparent volume of distribution (Vz/F) of the first dose ranged from 50.5 L to 92 L and was generally consistent across the dose groups from 3.2 mg to 32 mg. The analogous median apparent volume of distribution at steady-state (Vz/Fss) was also generally consistent and ranged from 43.2 L to 121 L.
Biotransformation: In vitro, cytochrome P450 3A4 was the predominant (>80%) isoform involved in the P450-mediated metabolism of lenvatinib. However, in vivo data indicated that non-P450-mediated pathways contributed to a significant portion of the overall metabolism of lenvatinib. Consequently, in vivo, inducers and inhibitors of CYP 3A4 had a minimal effect on lenvatinib exposure (see Interactions).
In human liver microsomes, the demethylated form of lenvatinib (M2) was identified as the main metabolite. M2' and M3', the major metabolites in human faeces, were formed from M2 and lenvatinib, respectively, by aldehyde oxidase.
In plasma samples collected up to 24 hours after administration, lenvatinib constituted 97% of the radioactivity in plasma radiochromatograms while the M2 metabolite accounted for an additional 2.5%. Based on AUC(0 - inf), lenvatinib accounted for 60% and 64% of the total radioactivity in plasma and blood, respectively.
Data from a human mass balance/excretion study indicate lenvatinib is extensively metabolised in humans. The main metabolic pathways in humans were identified as oxidation by aldehyde oxidase, demethylation via CYP3A4, glutathione conjugation with elimination of the O-aryl group (chlorophenyl moiety), and combinations of these pathways followed by further biotransformations (e.g., glucuronidation, hydrolysis of the glutathione moiety, degradation of the cysteine moiety, and intramolecular rearrangement of the cysteinylglycine and cysteine conjugates with subsequent dimerisation). These in vivo metabolic routes align with the data provided in the in vitro studies using human biomaterials.
In vitro transporter studies: For the following transporters, OAT1, OAT3, OATP1B1, OCT1, OCT2, and BSEP, clinically relevant inhibition was excluded based on a cutoff of IC50> 50 x Cmax.unbound.
Lenvatinib showed minimal or no inhibitory activities toward P-gp-mediated and BCRP-mediated transport activities. Similarly, no induction of P-gp mRNA expression was observed.
Lenvatinib showed minimal or no inhibitory effect on OATP1B3 and MATE2-K. Lenvatinib weakly inhibits MATE1. In human liver cytosol, lenvatinib did not inhibit aldehyde oxidase activity.
Elimination: Plasma concentrations decline bi-exponentially following Cmax. The mean terminal exponential half-life of lenvatinib is approximately 28 hours.
Following administration of radiolabelled lenvatinib to 6 patients with solid tumours, approximately two-thirds and one-quarter of the radiolabel were eliminated in the faeces and urine, respectively. The M3 metabolite was the predominant analyte in excreta (~17% of the dose), followed by M2' (~11% of the dose) and M2 (~4.4% of the dose).
Linearity/non-linearity: Dose proportionality and accumulation: In patients with solid tumours administered single and multiple doses of lenvatinib once daily, exposure to lenvatinib (Cmax and AUC) increased in direct proportion to the administered dose over the range of 3.2 to 32 mg once-daily (QD).
Lenvatinib displays minimal accumulation at steady state. Over this range, the median accumulation index (Rac) ranged from 0.96 (20 mg) to 1.54 (6.4 mg). The Rac in HCC subjects with mild and moderate liver impairment was similar to that reported for other solid tumours.
Special populations: Hepatic impairment: The pharmacokinetics of lenvatinib following a single 10-mg dose were evaluated in 6 subjects each with mild and moderate hepatic impairment (Child-Pugh A and Child-Pugh B, respectively). A 5-mg dose was evaluated in 6 subjects with severe hepatic impairment (Child-Pugh C). Eight healthy, demographically matched subjects served as controls and received a 10-mg dose. Lenvatinib exposure, based on dose-adjusted AUC0-t and AUC0-inf data, was 119%, 107%, and 180% of normal for subjects with mild, moderate, and severe hepatic impairment, respectively. It has been determined that plasma protein binding in plasma from hepatically impaired subjects was similar to the respective matched healthy subjects and no concentration dependency was observed. See Dosage & Administration for dosing recommendation.
There are not sufficient data for HCC patients with Child-Pugh B (moderate hepatic impairment, 3 patients treated with lenvima in the pivotal trial) and no data available in Child Pugh C HCC patients (severe hepatic impairment). Lenvatinib is mainly eliminated via the liver and exposure might be increased in these patient populations.
The median half-life was comparable in subjects with mild, moderate, and severe hepatic impairment as well as those with normal hepatic function and ranged from 26 hours to 31 hours. The percentage of the dose of lenvatinib excreted in urine was low in all cohorts (<2.16% across treatment cohorts).
Renal impairment: The pharmacokinetics of lenvatinib following a single 24-mg dose were evaluated in 6 subjects each with mild, moderate, and severe renal impairment, and compared with 8 healthy, demographically matched subjects. Subjects with end-stage renal disease were not studied.
Lenvatinib exposure, based on AUC0-inf data was 101%, 90%, and 122% of normal for subjects with mild, moderate, and severe renal impairment, respectively. It has been determined that plasma protein binding in plasma from renally impaired subjects was similar to the respective matched healthy subjects and no concentration dependency was observed. See Dosage & Administration for dosing recommendation.
Age, sex, weight, race, tumor type: Based on a population pharmacokinetic analysis of patients receiving up to 24 mg lenvatinib once daily as monotherapy (DTC), up to 18 mg once daily in combination with 5 mg everolimus (RCC), and up to 20 mg once daily in combination with pembrolizumab (RCC and EC), age, sex, weight, race (Japanese vs. other, Caucasian vs. other) and tumor type had no significant effects on clearance (see Dosage & Administration).
Paediatric Population: Paediatric patients have not been studied.
Toxicology: Preclinical safety data: In the repeated-dose toxicity studies (up to 39 weeks), lenvatinib caused toxicologic changes in various organs and tissues related to the expected pharmacologic effects of lenvatinib including glomerulopathy, testicular hypocellularity, ovarian follicular atresia, gastrointestinal changes, bone changes, changes to the adrenals (rats and dogs) and arterial (arterial fibrinoid necrosis, medial degeneration, or haemorrhage) lesions in rats, dogs, and cynomolgus monkeys. Elevated transaminase levels associated with signs of hepatotoxicity, were also observed in rats, dogs and monkeys. Reversibility of the toxicologic changes was observed at the end of a 4-week recovery period in all animal species investigated.
Genotoxicity: Lenvatinib mesilate was not mutagenic in the in vitro bacterial reverse mutation (Ames) assay. Lenvatinib was not clastogenic in the in vitro mouse lymphoma thymidine kinase assay or the in vivo micronucleus assay in rats.
Carcinogenicity: Carcinogenicity studies have not been conducted with lenvatinib.
Reproductive and developmental toxicity: No specific studies with lenvatinib have been conducted in animals to evaluate the effect on fertility. However, testicular (hypocellularity of the seminiferous epithelium) and ovarian changes (follicular atresia) were observed in repeated-dose toxicity studies in animals at exposures 11 to 15 times (rat) or 0.6 to 7 times (monkey) the anticipated clinical exposure (based on AUC) at the maximum tolerated human dose. These findings were reversible at the end of a 4-week recovery period.
Administration of lenvatinib during organogenesis resulted in embryolethality and teratogenicity in both rats (foetal external and skeletal anomalies) at exposures below the clinical exposure (based on AUC) at the maximum tolerated human dose, and rabbits (foetal external, visceral or skeletal anomalies) based on body surface area; mg/m2 at the maximum tolerated human dose. These findings indicate that lenvatinib has a teratogenic potential, likely related to the pharmacologic activity of lenvatinib as an antiangiogenic agent. Lenvatinib and its metabolites are excreted in rat milk.
Juvenile animal toxicity studies: Mortality was the dose-limiting toxicity in juvenile rats in which dosing was initiated on postnatal day (PND) 7 or PND21 and was observed at exposures that were respectively 125- or 12-fold lower compared with the exposure at which mortality was observed in adult rats, suggesting an increasing sensitivity to toxicity with decreasing age. Therefore, mortality may be attributed to complications related to primary duodenal lesions with possible contribution from additional toxicities in immature target organs.
The toxicity of lenvatinib was more prominent in younger rats (dosing initiated on PND 7) compared with those with dosing initiated on PND21 and mortality and some toxicities were observed earlier in the juvenile rats at 10 mg/kg compared with adult rats administered the same dose level. Growth retardation, secondary delay of physical development, and lesions attributable to pharmacologic effects (incisors, femur [epiphyseal growth plate], kidneys, adrenals, and duodenum) were also observed in juvenile rats.