Pharmacotherapeutic group: Antineoplastic agents, HER2 (Human Epidermal Growth Factor Receptor 2) inhibitors.
ATC code: L01FD04.
Pharmacology: Pharmacodynamics: Mechanism of Action: ENHERTU, trastuzumab deruxtecan, is a HER2-targeted antibody-drug conjugate (ADC). The antibody is a humanized anti-HER2 IgG1 attached to deruxtecan, a topoisomerase I inhibitor bound by a tetrapeptide-based cleavable linker. The ADC is stable in plasma. Following binding to HER2 on tumor cells, trastuzumab deruxtecan undergoes internalization and intracellular linker cleavage by lysosomal enzymes that are upregulated in cancer cells. Upon release, the membrane-permeable topoisomerase I inhibitor causes DNA damage and apoptotic cell death. The topoisomerase I inhibitor, an exatecan derivative, is approximately 10 times more potent than SN-38, the active metabolite of irinotecan.
Pharmacodynamic Effects: The administration of multiple doses of trastuzumab deruxtecan (6.4 mg/kg every 3 weeks) did not show any clinically meaningful effect on the QTc interval in an open-label, single-arm study in 51 patients with HER2-expressing metastatic breast cancer.
Clinical Efficacy: Metastatic Breast Cancer: DESTINY-Breast03: The efficacy and safety of ENHERTU were demonstrated in a Phase 3, randomized, multicenter, open-label, active-controlled study: DESTINY-Breast03.
The study included adult patients with unresectable or metastatic HER2-positive breast cancer who received prior trastuzumab and taxane therapy for metastatic disease or developed disease recurrence during or within 6 months of completing adjuvant therapy. Archival breast tumor samples were required to show HER2 positivity defined as HER2 IHC 3+ or ISH-positive. The study excluded patients with a history of ILD/pneumonitis requiring treatment with steroids or ILD/pneumonitis at screening, patients with untreated or symptomatic brain metastases, patients with a history of clinically significant cardiac disease, and patients with prior treatment with an anti-HER2 antibody-drug conjugate in the metastatic setting. Patients were randomized 1:1 to receive either ENHERTU 5.4 mg/kg (N=261) or trastuzumab emtansine 3.6 mg/kg (N=263) by intravenous infusion every three weeks. Randomization was stratified by hormone receptor status, prior treatment with pertuzumab, and history of visceral disease. Treatment was administered until disease progression, death, withdrawal of consent, or unacceptable toxicity.
The primary efficacy outcome measure was progression-free survival (PFS) as assessed by blinded independent central review (BICR) based on RECIST v1.1. Overall survival (OS) was a key secondary efficacy outcome measure. PFS based on investigator assessment, confirmed objective response rate (ORR), duration of response (DOR), Patient-Reported Outcomes (PRO), and time to hospitalization were secondary endpoints.
Demographic and baseline disease characteristics were similar between treatment arms. Of the 524 patients randomized, the median age was 54 years (range 20 to 83); female (99.6%); Asian (59.9%), White (27.3%), Black or African American (3.6%); Eastern Cooperative Oncology Group (ECOG) performance status 0 (62.8%) or 1 (36.8%); hormone receptor status (positive: 51.9%); presence of visceral disease (73.3%); previously treated and stable brain metastases (21.8%), and 48.3% of patients received one line of prior systemic therapy in the metastatic setting. The percentage of patients who had not received prior treatment for metastatic disease was 9.5%. The most common prior anti-HER2 cancer therapies received by patients included trastuzumab (99.6%), pertuzumab (61.1%), and an anti-HER2 tyrosine kinase inhibitor (14.9%).
At the prespecified interim analysis for PFS based on 245 events (73% of total events planned for final analysis), the study demonstrated a statistically significant improvement in PFS per BICR in patients randomized to ENHERTU compared to trastuzumab emtansine. Overall survival (OS) was immature at the time of analysis.
Efficacy results are summarized in Table 1 and Figures 1 and 2. (See Table 1 and Figures 1 and 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Similar PFS results were observed across prespecified subgroups including prior pertuzumab therapy, hormone receptor status, presence of stable brain metastases, and presence of visceral disease.
As secondary outcome measures, the PRO variables showed that the Quality of Life (QoL) of patients in the ENHERTU arm was either maintained or numerically improved on treatment compared with patients in the trastuzumab emtansine arm. The mean changes from baseline in European Organization for Research and Treatment of Cancer (EORTC) quality of life questionnaire (QLQ)-C30 global health status (the primary PRO variable) demonstrated that overall health and QoL were maintained while patients were on treatment with ENHERTU. The PRO results should be interpreted in the context of an open-label study design and therefore taken cautiously.
For all prespecified subscales, the hazard ratio (HR) for time to definitive deterioration numerically favored the ENHERTU arm over the trastuzumab emtansine arm (HR ranging from 0.69 to 0.90). Median time to definitive deterioration for the global health status from the EORTC QLQ-C30 was 9.7 months (95% CI: 7.3, 12.5) for the ENHERTU arm and 8.3 months (95% CI: 7.0, 10.3) for the trastuzumab emtansine arm (HR: 0.88 [95% CI: 0.70, 1.11]). The unadjusted p-values for the time to definitive deterioration HRs were less than 0.05 for EORTC QLQ-C30 emotional functioning (HR 0.69 [95% CI 0.53, 0.89]; p-value = 0.0049) and pain symptoms (HR 0.75 [95% CI: 0.59, 0.95]; p-value = 0.0146) subscales, as well as for the visual analogue scale of the EuroQoL-5 dimensions-5 levels of severity (EQ-5D-5L) (HR 0.77 [95% CI: 0.61, 0.98]; p-value = 0.0354) and the arm symptoms subscale of the EORTC QLQ-BR45 (HR 0.70 [95% CI: 0.55, 0.89]; p-value = 0.0033).
Among the 18 (6.9%) patients in the ENHERTU arm and the 19 (7.2%) patients in the trastuzumab emtansine arm who were hospitalized, time to first hospitalization was longer in the ENHERTU arm (median of 219.5 days and 60.0 days, respectively).
DESTINY-Breast01: The efficacy and safety of ENHERTU were demonstrated in a Phase 2, single-agent, open-label, multicenter study: DESTINY-Breast01.
The study included adult patients with unresectable or metastatic HER2-positive breast cancer who had received two or more prior anti-HER2 regimens, including trastuzumab emtansine (100%), trastuzumab (100%), and pertuzumab (65.8%). Archival breast tumor samples were required to show HER2 positivity defined as HER2 IHC 3+ or ISH-positive. The study excluded patients with a history of treated ILD or ILD at screening and patients with a history of clinically significant cardiac disease. ENHERTU was administered by intravenous infusion at 5.4 mg/kg once every three weeks until disease progression, death, withdrawal of consent, or unacceptable toxicity. The primary efficacy outcome measure was confirmed objective response rate (ORR) according to Response Evaluation Criteria in Solid Tumors (RECIST v1.1) in the intent-to-treat (ITT) population as evaluated by independent central review (ICR). Duration of response (DOR) and progression-free survival (PFS) were additional outcome measures.
DESTINY-Breast01 (N=184) baseline demographic and disease characteristics were: median age 55 years (range 28 to 96); female (100%); White (54.9%), Asian (38.0%), Black or African American (2.2%); Eastern Cooperative Oncology Group (ECOG) performance status 0 (55.4%) or 1 (44.0%); hormone receptor status (positive: 52.7%); presence of visceral disease (91.8%); median number of prior therapies in the metastatic setting: 5 (range: 2 to 17); prior pertuzumab therapy (65.8%); sum of diameters of target lesions (<5 cm: 42.4%, ≥5 cm: 50.0%).
Efficacy results are summarized in Table 2. (See Table 2.)
Click on icon to see table/diagram/image
Consistent antitumor activity was observed with ENHERTU regardless of prior pertuzumab therapy and hormone receptor status. In DESTINY-Breast01, the subgroup of patients who received prior pertuzumab therapy had a confirmed ORR of 66% (95% CI: 57, 76), and those who did not receive prior pertuzumab therapy had a confirmed ORR of 55% (95% CI: 42, 68). The subgroup of patients who were hormone receptor positive at baseline had a confirmed ORR of 59% (95% CI: 48, 69), and those who were HR- at baseline had a confirmed ORR of 68% (95% CI: 56, 77).
DESTINY-Breast04: The efficacy and safety of ENHERTU were evaluated in study DESTINY-Breast04, a Phase 3, randomized, multicenter, open-label study that enrolled 557 adult patients with unresectable or metastatic HER2-low breast cancer. The study included 2 cohorts: 494 hormone receptor positive (HR+) patients and 63 hormone receptor negative (HR-) patients. HER2-low expression was defined as IHC 1+ or IHC 2+/ISH-, as determined by the PATHWAY/VENTANA anti-HER-2/neu (4B5) evaluated at a central laboratory. Patients must have received chemotherapy in the metastatic setting or have developed disease recurrence during or within 6 months of completing adjuvant chemotherapy. Patients who were HR+ must have received at least one endocrine therapy or be ineligible for endocrine therapy. Patients were randomized 2:1 to receive either ENHERTU 5.4 mg/kg (N=373) by intravenous infusion every three weeks or physician's choice of chemotherapy (N=184, eribulin 51.1%, capecitabine 20.1%, gemcitabine 10.3%, nab paclitaxel 10.3%, or paclitaxel 8.2%). Randomization was stratified by HER2 IHC status of tumor samples (IHC 1+ or IHC 2+/ISH-), number of prior lines of chemotherapy in the metastatic setting (1 or 2), and HR status/prior CDK4/6i treatment (HR+ with prior CDK4/6 inhibitor treatment, HR+ without prior CDK4/6 inhibitor treatment, or HR-). Treatment was administered until disease progression, death, withdrawal of consent, or unacceptable toxicity. The study excluded patients with a history of ILD/pneumonitis requiring treatment with steroids or ILD/pneumonitis at screening and clinically significant cardiac disease. Patients were also excluded for untreated or symptomatic brain metastases or ECOG performance status >1.
The primary efficacy outcome measure was PFS in patients with HR+ breast cancer assessed by BICR based on RECIST v1.1. Key secondary efficacy outcome measures were PFS assessed by BICR based on RECIST v1.1 in the overall population (all randomized HR+ and HR- patients), OS in HR+ patients, and OS in the overall population. ORR, DOR, and PROs were secondary endpoints.
Demographics and baseline tumor characteristics were similar between treatment arms. Of the 557 patients randomized, the median age was 56.5 years (range: 28.4 to 80.5); 23.5% were age 65 or older; 99.6% were female and 0.4% were male; 47.9% were White, 40.0% were Asian, and 1.8% were Black or African American. Patients had an ECOG performance status of 0 (54.8%) or 1 (45.2%) at baseline; 57.6% were IHC 1+, 42.4% were IHC 2+/ISH-; 69.8% had liver metastases, 32.9% had lung metastases, and 5.7% had brain metastases. In the metastatic setting, patients had a median of 3 prior lines of systemic therapy (range: 1 to 9) with 57.6% having 1 and 40.9% having 2 prior chemotherapy regimens; 3.9% were early progressors (progression in the neo/adjuvant setting). In HR+ patients, the median number of prior lines of endocrine therapy was 2 (range: 0 to 9) and 70% had prior CDK4/6i treatment.
The study demonstrated a statistically significant and clinically meaningful improvement in OS and PFS in patients randomized to ENHERTU compared to chemotherapy in both the HR+ cohort and the overall population.
Efficacy results are summarized in Table 3 and Figures 3 and 4. (See Table 3, Figures 3 and 4.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Locally Advanced or Metastatic Gastric Cancer: The efficacy and safety of ENHERTU were demonstrated in a Phase 2, multicenter, open-label, randomized study: DESTINY-Gastric01. The study included adult patients with locally advanced or metastatic HER2-positive gastric or GEJ adenocarcinoma who had progressed on at least two prior regimens, including trastuzumab, a fluoropyrimidine agent, and a platinum agent. Patients were randomized 2:1 to receive either ENHERTU (N=126) or physician's choice of chemotherapy: either irinotecan (N=55) or paclitaxel (N=7). Randomization was stratified by HER2 status (IHC 3+ or IHC 2+/ISH-positive), ECOG performance status (0 or 1), and region (Japan or South Korea). ENHERTU was administered by intravenous infusion at 6.4 mg/kg every three weeks. Irinotecan monotherapy was administered by intravenous infusion biweekly at 150 mg/m
2. Paclitaxel monotherapy was administered by intravenous infusion weekly at 80 mg/m
2. Tumor samples were required to have centrally confirmed HER2 positivity defined as IHC 3+ or IHC 2+/ISH-positive. The study excluded patients with a history of treated ILD and/or ILD at screening, patients with a history of clinically significant cardiac disease, and patients with active brain metastases. Treatment was administered until disease progression, death, withdrawal of consent, or unacceptable toxicity. The primary efficacy outcome measure was ORR assessed by ICR based on RECIST v1.1. Overall survival (OS) was a key secondary endpoint. PFS, DOR, and confirmed ORR were additional secondary outcome measures.
Demographic and baseline disease characteristics were similar between treatment arms. Of the 188 patients, the median age was 66 years (range 28 to 82); 76% were male; 100% were Asian. Patients had an ECOG performance status of either 0 (49%) or 1 (51%); 87% had gastric adenocarcinoma and 13% had GEJ adenocarcinoma; 76% were IHC 3+ and 23% were IHC 2+/ISH-positive; 65% had inoperable advanced cancer; 35% had postoperative recurrent cancer; 54% had liver metastases; 29% had lung metastases; the sum of diameters of target lesions was <5 cm in 47%, ≥5 to <10 cm in 30%, and ≥10 cm in 17%; 55 % had two and 45% had three or more prior regimens in the locally advanced or metastatic setting.
The study demonstrated a statistically significant and clinically meaningful improvement in ORR and OS in the ENHERTU-treated group compared to the chemotherapy-treated group.
Efficacy results are summarized in Table 4 and the Kaplan-Meier curve for OS is shown in Figure 5. (See Table 4 and Figure 5.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In the exploratory subgroup analysis of patients who were HER2 IHC 3+, confirmed ORR was 46.9% for Enhertu (N=96; 95% CI: 36.6, 57.3), and 8.5% for chemotherapy (N=47; 95% CI: 2.4, 20.4). In the subgroup of patients who were IHC 2+/ISH-positive, confirmed ORR was 20.7% for Enhertu (N=29; 95% CI: 8.0, 39.7), and 20.0% for chemotherapy (N=15; 95% CI: 4.3, 48.1). In the subgroup of patients who were IHC 3+, median OS was 12.8 months for Enhertu (N=96; 95% CI: 10.3, 18.0) and 8.6 months for chemotherapy (N=47; 95% CI: 6.4, 10.7). In the subgroup of patients who were IHC 2+/ISH-positive, median OS was 10.1 months for Enhertu (N=29; 95% CI: 5.4, 13.1) and 8.4 months for chemotherapy (N=15; 95% CI 3.9, 20.0). The number of IHC 2+/ISH-positive patients was small which limits drawing any meaningful conclusions.
Pharmacokinetics: Distribution: Based on population pharmacokinetic analysis, the volume of distribution of the central compartment (Vc) of trastuzumab deruxtecan was estimated to be 2.68 L.
In vitro, the mean human plasma protein binding of the topoisomerase I inhibitor was approximately 97%.
In vitro, the blood-to-plasma concentration ratio of the topoisomerase I inhibitor was approximately 0.6.
Biotransformation: Trastuzumab deruxtecan undergoes intracellular cleavage by lysosomal enzymes to release the active topoisomerase I inhibitor.
The humanized HER2 IgG1 monoclonal antibody is expected to be degraded into small peptides and amino acids via catabolic pathways in the same manner as endogenous IgG.
In vitro metabolism studies in human liver microsomes indicate that the topoisomerase I inhibitor is metabolized mainly by CYP3A4 via oxidative pathways.
Elimination: Based on population pharmacokinetic analysis, following intravenous administration of trastuzumab deruxtecan in patients with metastatic HER2-positive or HER2-low breast cancer and locally advanced or metastatic gastric or GEJ adenocarcinoma, the clearance of trastuzumab deruxtecan was estimated to be 0.41 L/day and the clearance of the topoisomerase I inhibitor was 19.6 L/h. The apparent elimination half-life (t
1/2) of trastuzumab deruxtecan was 5.7-5.8 days and of released topoisomerase I inhibitor was approximately 5.5-5.8 days. In vitro, topoisomerase I inhibitor was a substrate of P-gp, OATP1B1, OATP1B3, MATE2-K, MRP1, and BCRP. Moderate accumulation of trastuzumab deruxtecan was observed at the 5.4 mg/kg and 6.4 mg/kg doses (approximately 35%-39% in cycle 3 compared to cycle 1).
Following intravenous administration of the topoisomerase I inhibitor to rats, the major excretion pathway was feces via the biliary route. The topoisomerase I inhibitor was the most abundant component in urine, feces, and bile. Following single intravenous administration of trastuzumab deruxtecan (6.4 mg/kg) to monkeys, unchanged released topoisomerase I inhibitor was the most abundant component in urine and feces.
Linearity/Nonlinearity: The exposure of trastuzumab deruxtecan and released topoisomerase I inhibitor when administered intravenously increased in proportion to dose in the 3.2 mg/kg to 8.0 mg/kg dose range (approximately 0.6 to 1.5 times the recommended dose) with low to moderate interindividual variability.
Pharmacokinetics in Special Populations: Based on population pharmacokinetic analysis, age (20-96 years), race, ethnicity, sex and body weight did not have a clinically meaningful effect on exposure of trastuzumab deruxtecan or released topoisomerase I inhibitor.
Renal: No dedicated renal impairment study was conducted. Based on population pharmacokinetic analysis including patients with mild (creatinine clearance [CL
cr] ≥60 and <90 mL/min) or moderate (CL
cr ≥30 and <60 mL/min) renal impairment (estimated by Cockcroft-Gault), the pharmacokinetics of the released topoisomerase I inhibitor was not affected by mild to moderate renal impairment as compared to normal renal function (CL
cr ≥90 mL/min).
Hepatic: No dedicated hepatic impairment study was conducted. Based on population pharmacokinetic analysis, higher levels of AST and total bilirubin resulted in a lower clearance of topoisomerase I inhibitor. The impact of these changes in patients with mild hepatic impairment is not expected to be clinically meaningful. The pharmacokinetics of trastuzumab deruxtecan or the topoisomerase I inhibitor in patients with moderate to severe hepatic impairment (total bilirubin >1.5 ULN with any AST) is unknown.
Drug Interaction Studies: Effects of Other Medicinal Products on the Pharmacokinetics of Trastuzumab Deruxtecan: In vitro studies indicate that the topoisomerase I inhibitor is metabolized mainly by CYP3A4 and is a substrate of the following transporters: P-gp, OATP1B1, OATP1B3, MATE2-K, MRP1, and BCRP.
Coadministration of ritonavir (200 mg twice daily from day 17 of cycle 2 to day 21 of cycle 3), a dual inhibitor of OATP1B/CYP3A, increased exposure (AUC) of trastuzumab deruxtecan by 19% and the released topoisomerase I inhibitor by 22%.
Coadministration of itraconazole (200 mg twice daily from day 17 of cycle 2 to day 21 of cycle 3), a strong CYP3A inhibitor, increased exposure (AUC) of trastuzumab deruxtecan by 11% and the released topoisomerase I inhibitor by 18%. The impact of these changes is not expected to be clinically meaningful.
No clinically relevant drug-drug interaction is expected with drugs that are inhibitors of P-gp, MATE2-K, MRP1, or BCRP transporters.
Effects of Trastuzumab Deruxtecan on the Pharmacokinetics of Other Medicinal Products: In vitro studies indicate that the topoisomerase I inhibitor does not inhibit or induce major CYP450 enzymes, including CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A. In vitro studies indicate that the topoisomerase I inhibitor does not inhibit OAT3, OCT1, OCT2, OATP1B3, MATE1, MATE2-K, P-gp, BCRP, or BSEP transporters, but has an inhibitory effect on OAT1 and OATP1B1 with IC
50 values of 12.7 and 14.4 µmol/L, respectively, which are significantly higher than steady-state C
max (0.02 µmol/L) of topoisomerase I inhibitor at 5.4 mg/kg dose administered every 3 weeks. No clinically meaningful drug-drug interaction is expected with drugs that are substrates of OAT1 or OATP1B1 transporters.
Toxicology: Nonclinical safety data: Animal Toxicology and/or Pharmacology: In a six-week repeat-dose toxicity study, trastuzumab deruxtecan was administered to rats once every three weeks at doses up to 197 mg/kg (approximately 31 times the clinical dose of 5.4 mg/kg based on AUC). Toxicities were observed in intestines, lymphatic/hematopoietic organs (thymus, lymph nodes, bone marrow), kidneys, skin, testes, and incisor teeth. All changes observed, except for testicular and incisor teeth changes, were reversible following a nine-week recovery period.
In a three-month repeat-dose toxicity study, trastuzumab deruxtecan was administered to monkeys once every three weeks at doses up to 30 mg/kg (approximately 9 times the clinical dose of 5.4 mg/kg based on AUC). Toxicities were observed in intestines, testes, skin, bone marrow, kidneys, and lungs. Pulmonary toxicity was observed at the highest dose (30 mg/kg) and histopathologically characterized by aggregation of foamy alveolar macrophages and focal alveolus and/or interstitial inflammation which showed reversibility after a three-month recovery period. Changes observed in other organs, except for those in the skin and kidney, also showed reversibility or a trend toward reversibility by the end of a three-month recovery period.
Mutagenesis/Carcinogenesis: The topoisomerase I inhibitor component of trastuzumab deruxtecan was clastogenic in both an in vivo rat bone marrow micronucleus assay and an in vitro Chinese hamster lung chromosome aberration assay and was not mutagenic in an in vitro bacterial reverse mutation assay.
Carcinogenicity studies have not been conducted with trastuzumab deruxtecan.
Impairment of Fertility and Teratogenicity: Dedicated fertility studies have not been conducted with trastuzumab deruxtecan. Based on results from general animal toxicity studies, trastuzumab deruxtecan may impair male reproductive function and fertility.
There were no animal reproductive or developmental toxicity studies conducted with trastuzumab deruxtecan. Based on results from general animal toxicity studies, trastuzumab deruxtecan and the topoisomerase I inhibitor component were toxic to rapidly dividing cells (lymphatic/hematopoietic organs, intestine, or testes), and the topoisomerase I inhibitor was genotoxic, suggesting the potential for embryotoxicity and teratogenicity.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image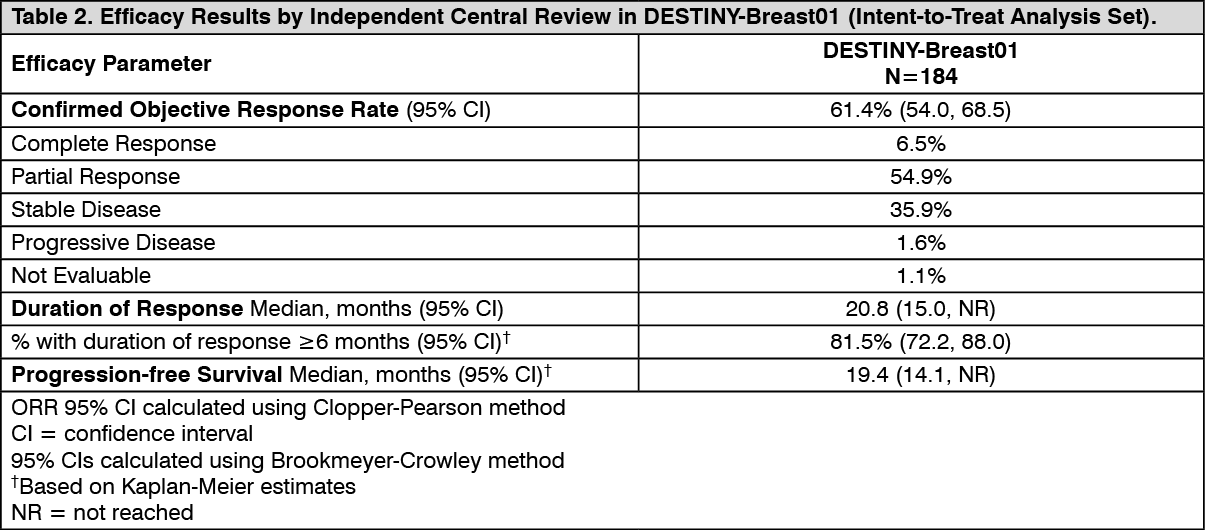 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
