


 Sign Out
Sign Out



Pharmacology: Pharmacodynamics: Clostridium botulinum type A toxin-haemagglutinin complex blocks peripheral cholinergic transmission at the neuromuscular junction by a presynaptic action at a site proximal to the release of acetylcholine.
The toxin acts within the nerve ending to antagonise those events that are triggered by Ca2+ which culminate in transmitter release. It does not affect postganglionic cholinergic transmission or postganglionic sympathetic transmission.
The action of toxin involves an initial binding step whereby the toxin attaches rapidly and avidly to the presynaptic nerve membrane. Secondly, there is an internalisation step in which toxin crosses the presynaptic membrane, without causing onset of paralysis. Finally the toxin inhibits the release of acetylcholine by disrupting the Ca2+ mediated acetylcholine release mechanism, thereby diminishing the endplate potential and causing paralysis.
Recovery of impulse transmission occurs gradually as new nerve terminals sprout and contact is made with the post synaptic motor endplate, a process which takes 6 - 8 weeks in the experimental animal.
Focal spasticity in adults: Upper limb: The efficacy and safety of Dysport for the treatment of upper limb spasticity was evaluated in a randomized, multi-centre, double-blind, placebo-controlled study that included 238 patients (159 Dysport and 79 placebo) with upper limb spasticity who were at least 6 months post-stroke (90%) or post-traumatic brain injury (10%). The primary targeted muscle group (PTMG) was the extrinsic finger flexors (56%), followed by the elbow (28%) and wrist flexors (16%).
The primary efficacy variable was the PTMG muscle tone at week 4, as measured by the Modified Ashworth Scale (MAS), a 5 point scale ranging from 0 (no increase in muscle tone) to 4 (affected in part[s] rigid in flexion or extension) and the first secondary endpoint was the Physician Global Assessment (PGA) of response to treatment (a 9 point scale ranging from -4 [markedly worse], through 0 [no change], to +4 [markedly improved]). The main results achieved at Week 4 and Week 12 are shown as follows: (See Table 1.)
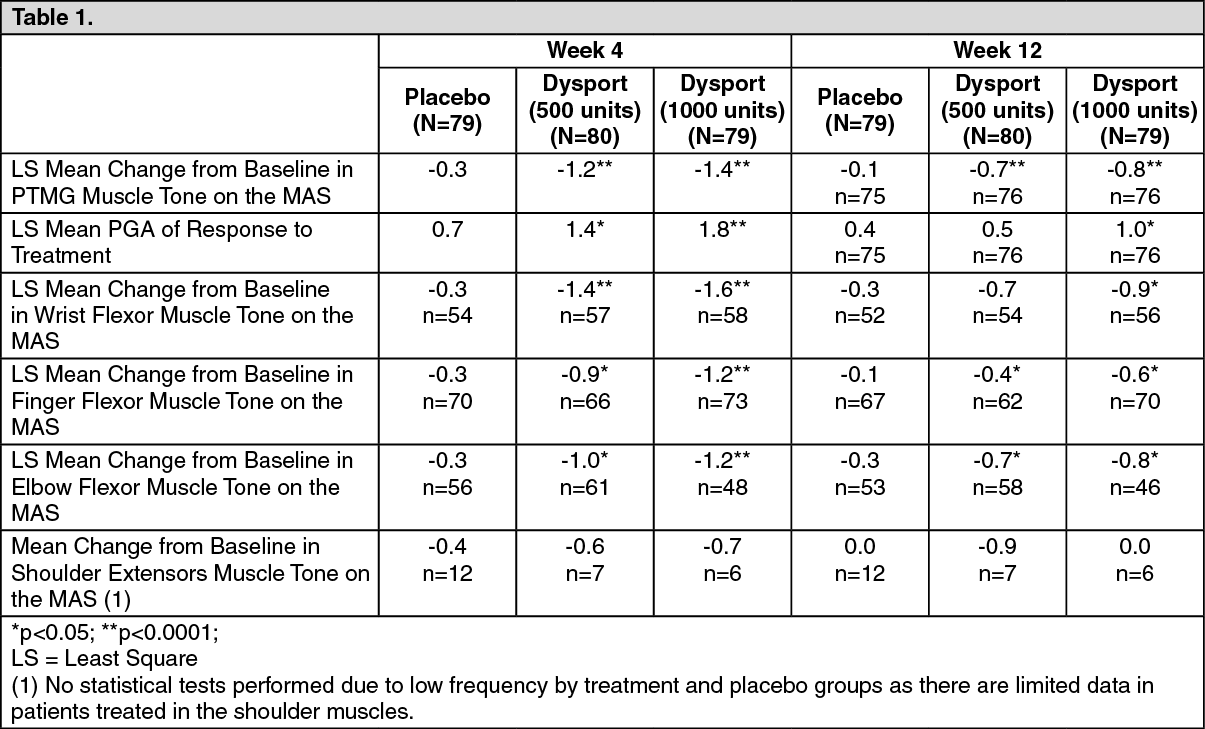 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe Principal Target of Treatment (PTT) of the Disability Assessment Scale [DAS] was used to investigate the effect of treatment on functional impairment (passive function). Although some improvement in the mean change from baseline at Week 4 in the Dysport groups did not reach statistical significance compared to placebo, the proportion of DAS score responders (subjects achieving at least a one grade improvement) for the PTT was significantly higher at the 1000U dose as shown as follows. (See Table 2.)
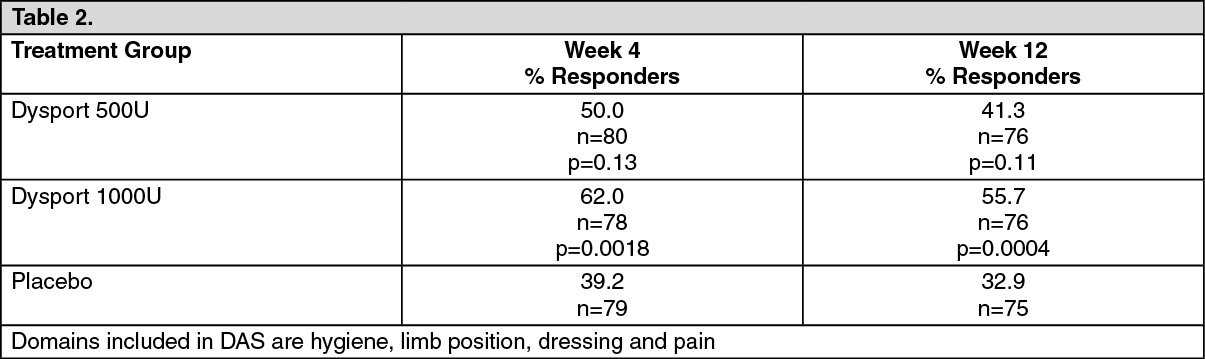 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn addition, statistically significant improvements in spasticity (grade and angle) assessed by the Tardieu scale, in the active range of motion of the fingers, wrist or elbow, and in ease of applying a splint by the subject were observed, especially at the 1000U dose. However, there was no effect of treatment shown on the active function, as assessed by the Modified Frenchay Score, and on quality of life EQ5D or SF-36 questionnaires.
Lower limb affecting the ankle joint: The efficacy and safety of Dysport for the treatment of lower limb spasticity was evaluated in a pivotal randomised, multi-centre, double-blind, placebo-controlled study that included 385 post-stroke and brain injury patients (255 Dysport and 130 placebo-treated subjects) with lower limb spasticity primarily affecting the ankle joint. Two doses of Dysport were evaluated for efficacy; Dysport 1000U (N = 125), Dysport 1500U (N = 128) against Placebo (N =128). The primary target muscle group was the gastrocnemius - soleus complex (GSC). The primary end point was Modified Ashworth Scale (MAS) score assessed at the ankle joint (with the knee extended) at week 4.
Dysport was divided between the GSC and at least one other distal or proximal lower limb muscle according to clinical presentation.
When assessing the primary endpoint, MAS at the ankle with the knee extended (involving all plantar flexors), statistically significant improvement was observed for 1500U. When assessing MAS at the ankle with the knee flexed (involving all plantar flexors except the gastrocnemius), statistically significant improvement was observed for both 1000U and 1500U. (See Table 3.)
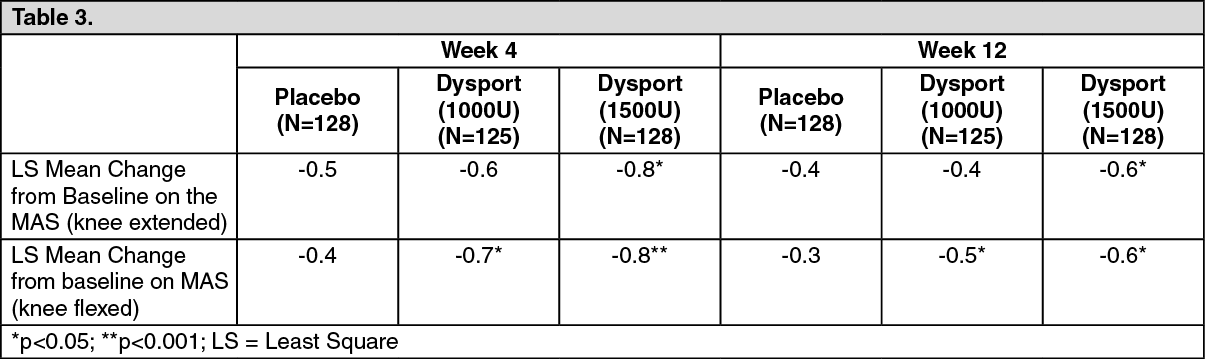 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSpasticity assessment using the Tardieu Scale (TS) showed that there were statistically significant improvements in spasticity grade at Weeks 4 to 20 in the Dysport 1500U group and at Weeks 4 to 12 in the Dysport 1000U group. In addition it showed statistically significant differences in Angle of Catch at Week 1 and 16, favouring the higher dose of Dysport.
Based on post hoc analysis due to non-normality of PGA data, Dysport treatment was also associated with statistically significant clinical improvement at both doses as measured by the Physician Global Assessment (PGA) Score.
Numerical improvement in ankle dorsiflexion for the higher Dysport dose was seen with the change peaking at 4 weeks post administration.
Additional endpoints such as reduction in pain, using walking aids and quality of life measures did not show statistically significant improvement.
On completion of this study, 345 patients entered an open-label extension study in which re-treatment with Dysport 1000U or 1500U was determined by clinical need.
This long term follow up study confirmed a prolonged treatment effect on spasticity related outcome measures following repeated injections.
Improvements in efficacy parameters (MAS, PGA and TS) seen after 4 weeks of double blind treatment with Dysport in the lower limb were maintained over repeated treatment.
Improvements in 10-m walking speed (comfortable and maximal, with or without shoes) were observed, which increased with successive treatment cycles.
No significant improvements in lower limb pain using the SPIN scale, use of walking aids or quality of life measures were observed.
Blepharospasm: Three Dysport doses were investigated over 1 treatment cycle in a clinical study.
Efficacy was measured by the medians of differences in the Percentage of Normal Activity (PNA) values (derived from the Blepharospasm Disability Scale) between each treatment group and placebo. A dose-dependent improvement in blepharospasm was evident with Dysport dose, with all treatment groups being superior to placebo. (See Table 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageFor the 40 units, 80 units and 120 units Dysport treatment groups, the medians of the changes from baseline in PNA values were statistically significantly higher compared to those in placebo group at weeks 4, 8, and 12.
A statistically significant difference compared to placebo group was also observed for the 80 units and 120 units Dysport treatment groups at week 16, indicating a greater duration of response at the 80 units and 120 units doses.
The incidence of related Treatment Emergent Adverse Events (TEAEs), specifically ptosis, was higher in the Dysport treatment groups than in the placebo treatment group and was dose-dependent with greater incidence seen at higher Dysport doses. (See Table 5 as follows.)
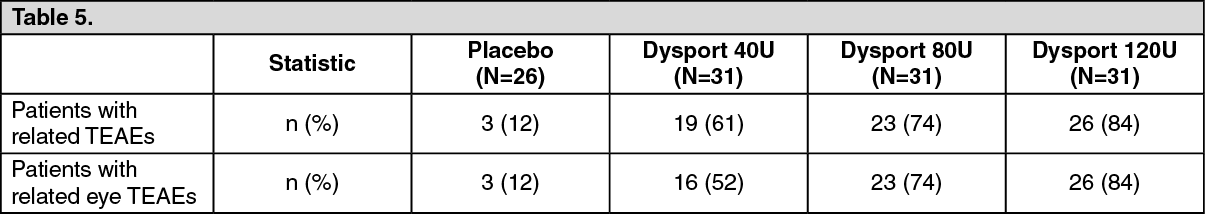 Click on icon to see table/diagram/image
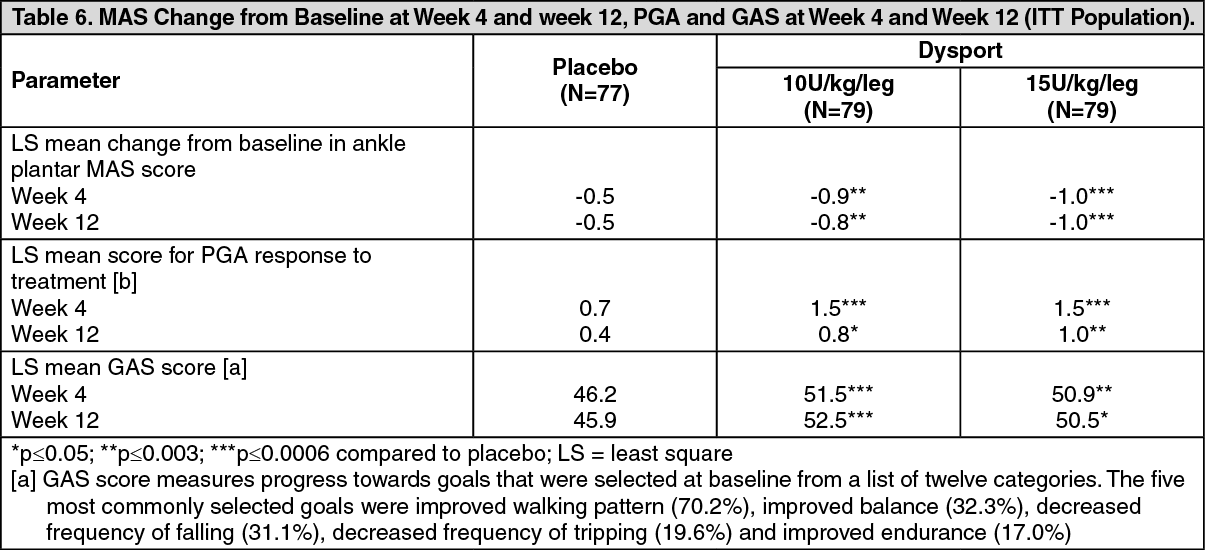
Click on icon to see table/diagram/imageFocal spasticity in paediatric cerebral palsy patients, two years of age or older: Dynamic equinus foot deformity due to focal spasticity in ambulant paediatric cerebral palsy patients, two years of age or older: A double-blind, placebo-controlled multicentre study (Study Y-55-52120-141) was conducted in children with dynamic equinus foot deformity due to spasticity in children with cerebral palsy. A total of 235 botulinum toxin naïve or non-naïve patients with a Modified Ashworth Score (MAS) of grade 2 or greater were enrolled to receive Dysport 10 units/kg/leg, Dysport 15 units/kg/leg or placebo. Forty one percent of patients were treated bilaterally resulting in a total Dysport dose of either 20 units/kg or 30 units/kg. The primary efficacy variable was the mean change from baseline in MAS in ankle plantar flexors at Week 4. Secondary efficacy variables were the mean Physicians Global Assessment (PGA) score and Mean Goal Attainment Scaling (GAS) score at Week 4. Patients were followed up for at least 12 weeks post-treatment and up to a maximum of 28 weeks. On completion of this study, patients were offered entry into an open-label extension study (Study Y-55-52120-147). (See Table 6.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageImprovement in the spasticity of the ankle plantar flexors was observed, as assessed by the Tardieu scale. The spasticity grade (Y) was statistically significantly improved compared to placebo for both the 10 units/kg/leg and 15 units/kg/leg Dysport treatment groups at Week 4 and Week 12, and the angle of catch (Xv3) was significant for the 10 units/kg/leg Dysport group at Week 12 and at both Week 4 and Week 12 for the 15 units/kg/leg Dysport group.
Both Dysport treatment groups, 10 units/kg/leg and 15 units/kg/leg, demonstrated a significant improvement from baseline in the Observational Gait Scale (OGS) overall score at Week 4 when compared to placebo and a statistically significantly higher proportion of patients were treatment responders for initial foot contact on the OGS at Week 4 and Week 12.
Parents completed the condition-specific Module for cerebral palsy for the Paediatric Quality of Life Inventory. There was a statistically significant improvement from baseline in fatigue at Week 12 in the Dysport 10 units/kg/leg and 15 units/kg/leg Dysport treatment groups compared to placebo. No other statistically significant improvements were observed in the other subscales.
On completion of this study, 216 patients entered an open-label extension study (Y-55-52120-147) where they could receive re-treatment based on clinical need. Both distal (gastrocnemius, soleus and tibialis posterior) and proximal (hamstrings and hip adductors) muscles were permitted to be injected, including multilevel injections. Efficacy was observed over repeated treatment sessions for up to 1 year as assessed by MAS, PGA and GAS.
Focal spasticity of upper limbs in paediatric cerebral palsy patients, two years of age or older: The efficacy and safety of Dysport for the treatment of upper limb spasticity in children was evaluated in a randomised, multi-centre, double-blind, controlled, study in which doses of 8 U/kg and 16 U/kg in the selected study upper limb were compared with a low dose control group of 2 U/kg. A total of 210 botulinum toxin naïve or non-naïve patients with upper limb spasticity due to cerebral palsy (Modified Ashworth Scale (MAS) score ≥2 in the primary targeted muscle group (PTMG)) were randomised and treated in the study.
The total dose of Dysport was injected intramuscularly into the affected upper limb muscles which included the PTMG of either elbow flexors or wrist flexors as well as other upper limb muscles according to the disease presentation. No more than 0.5 ml was allowed to be administered per injection site. However more than one injection site per muscle was permitted. An Electrical stimulation (ES) and/or ultrasound was used to assist muscle localization for injection.
After the initial treatment, up to 3 further treatments of Dysport could be administered at planned doses of either 8 U/kg or 16 U/kg, although the investigator could elect to increase or decrease the dose (but not exceeding 16 U/kg). The minimum retreatment interval was 16 weeks. For treatment cycles 2, 3 and 4, injection into the lower limbs and the non-study upper limb was also allowed at the same time as the study upper limb was injected. Subjects were followed-up for a minimum of 1 year to a maximum of 1 year 9 months after entry into the study.
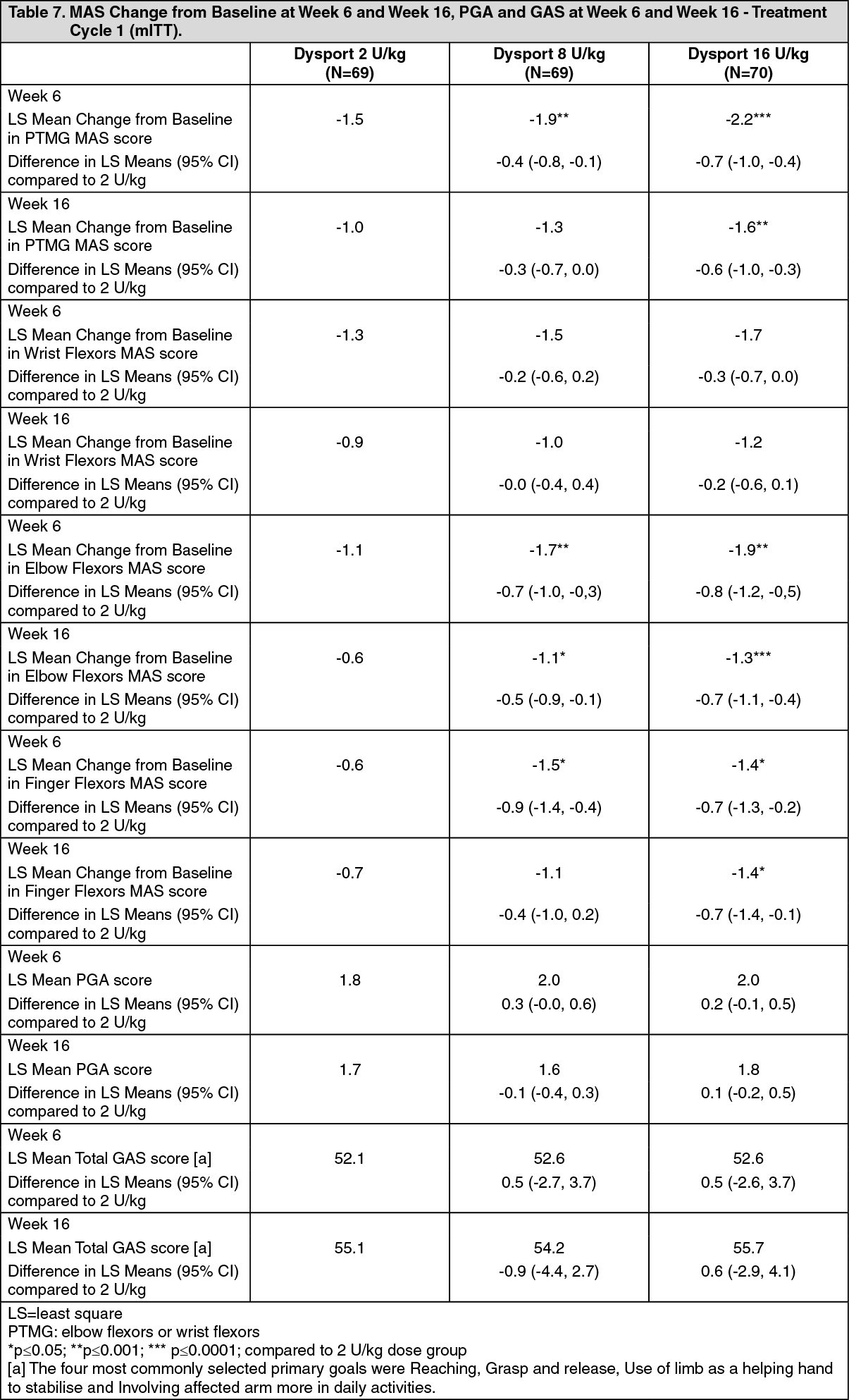
The primary efficacy variable was the mean change from baseline in MAS in PTMG at Week 6. Secondary efficacy variables were the mean Physicians Global Assessment (PGA) score and mean Goal Attainment Scale (GAS) score at Week 6. (See Table 7.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageImprovement in the spasticity of the PTMG was observed, as assessed by the Tardieu scale. In the PTMG elbow flexors, the angle of catch (Xv3) was statistically significantly improved compared with Dysport 2 U/kg at Week 6 for both the 8 and 16 U/kg treatment groups and also at Week 16 for the Dysport 16 U/kg group. In addition, a statistically significant decrease from Baseline in spasticity grade (Y) at Week 6 and 16 was observed for the Dysport 16 U/kg group compared with Dysport 2 U/kg. In the PTMG wrist flexors, statistically significant improvements from Baseline in Xv3 and Y were observed in the Dysport 16 U/kg group compared with the Dysport 2 U/kg group at Week 6 but not for the 8 U/kg group.
Parents completed the condition-specific Module for Cerebral Palsy for the Paediatric Quality of Life Inventory. At Week 16, there was a statistically significant improvement from Baseline in fatigue (p=0.0251) in the Dysport 8 U/kg group and, in movement and balance (p=0.0253) in the 16 U/kg group compared with the Dysport 2 U/kg group. No other statistically significant improvements were observed in the other subscales.
The majority of subjects treated with Dysport were retreated by Week 28 (62.3% in the Dysport 8 U/kg group and 61.4% in the Dysport 16 U/kg group), though more than 24% of subjects in both treatment groups had not yet required retreatment by Week 34.
Following repeated treatment, efficacy was generally maintained across treatment cycles for both Dysport 8 U/kg and 16 U/kg groups.
Moderate to severe glabellar lines and lateral canthal lines: During the clinical development of Dysport, for the treatment of moderate to severe glabellar lines and lateral canthal lines, more than 4500 patients were included in the different clinical trials and approximately 3800 patients were exposed to Dysport.
Glabellar lines: In clinical studies, 2032 patients with moderate to severe glabellar lines have been treated at the recommended dose of 50U of Dysport. Of these, 305 were treated with 50U in two pivotal Phase III double-blind placebo-controlled studies and 1200 treated with 50U in a long-term open-label repeated dose Phase III study. The remaining patients were treated in supportive and dose-ranging studies.
The median time to onset of response was 2 to 3 days following treatment, with the maximum effect observed at day thirty. In both pivotal placebo-controlled phase III studies, Dysport injections significantly reduced the severity of glabellar lines for up to 4 months. The effect was still significant after 5 months in one of the two pivotal studies.
Thirty days after injection, the assessment of the investigators showed that 90% (273/305) of patients had responded to treatment (exhibited no or mild glabellar lines at maximum frown), compared to 3% (4/153) placebo-treated patients. Five months after injection, 17% (32/190) of patients treated with Dysport were still responding to treatment compared to 1% (1/92) of placebo treated patients in the concerned study. The patients' own assessment at maximum frown after thirty days gave a response rate of 82% (251/305) for those treated with Dysport and 6% (9/153) for those treated with placebo. The proportion of patients exhibiting a two-grade improvement according to the investigator assessment at maximum frown, was 77% (79/103) in the one pivotal Phase III study where this was assessed.
A subset of 177 patients had moderate or severe glabellar lines at rest prior to treatment. Assessment by investigators of this population, thirty days after treatment, showed that 71% (125/177) of Dysport treated patients were considered responders versus 10% (8/78) of placebo treated patients.
The long-term repeat dose open-label study showed that the median time to onset of response of 3 days was maintained across repeated dose cycles. The responder rate at maximum frown as determined by the investigator at day 30 was maintained over repeated cycles (ranging between 80% and 91% over the 5 cycles). The responder rate at rest over repeated dose cycles was also consistent with the single dose studies, with 56% to 74% of Dysport treated patients considered by investigators to be responders thirty days after treatment.
Lateral canthal lines: In clinical studies, 308 patients with moderate to severe lateral canthal lines at maximum smile have been treated at the recommended dose of 30 units per side in double-blind studies. Of these, 252 were treated in a Phase III double-blind placebo controlled study and 56 patients were treated in a double-blind Phase II dose-ranging study.
In the phase III study, Dysport injections significantly reduced the severity of lateral canthal lines compared with placebo (p≤0.001) at 4, 8 and 12 weeks (assessed at maximum smile by the investigators). For the subjects' assessment of satisfaction with the appearance of their lateral canthal lines, there was a statistically significant difference between Dysport and placebo (p≤0.010) in favour of Dysport at 4, 8, 12 and 16 weeks.
The primary efficacy endpoint was at 4 weeks following injection: the assessment of the investigators showed that 47.2% (119/252) of patients had responded to treatment (exhibited no or mild lateral canthal lines at maximum smile), compared to 7.2% (6/83) placebo-treated patients.
In a post-hoc analysis, at the same time point, 4 weeks following injection, 75% (189/252) of Dysport treated patients had at least 1 grade improvement at maximum smile compared with only 19% (16/83) of placebo-treated subjects.
A total of 315 subjects entered the open label extension phase of the Phase III study in which they could be treated concomitantly for both lateral canthal lines and glabellar lines. Patients treated with Dysport in the double-blind and open label phases of the Phase III received a median of 3 treatments for lateral canthal lines. The median interval between injections for lateral canthal lines, which was largely determined by the protocol design, ranged from 85 to 108 days. The results showed that efficacy is maintained with repeated treatments over the period of one year.
The patient satisfaction levels at weeks 4, 16 and 52 show after the first treatment with Dysport that 165/252 subjects (65.5%) were either very satisfied or satisfied with the appearance of their LCLs.
At week 16, 4 weeks after either a second Dysport treatment for those randomised to Dysport in Part A or the first treatment for those randomised to placebo the proportion who were very satisfied or satisfied was 233/262 (89.0%). At week 52 when subjects could have had up to five cycles of Dysport treatment with the last one being at week 48 the proportion of very satisfied/satisfied subjects was 255/288 (84.7%).
No patient tested positive for toxin-neutralising antibodies after receiving repeated treatments with Dysport over one year.
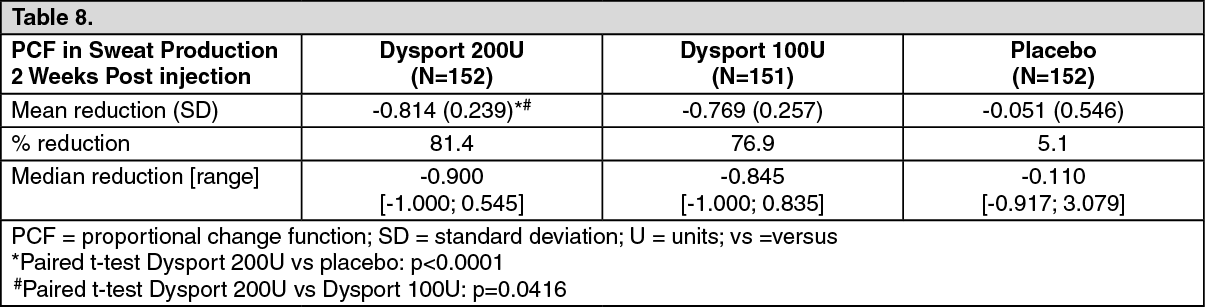
Axillary hyperhidrosis: The efficacy and safety of Dysport for the treatment of Axillary Hyperhidrosis was evaluated in a multi-centre, randomised, double-blind clinical study that included 152 adult patients with Axillary Hyperhidrosis who had symptoms for greater than one year and had failed standard therapy. Patients were injected with 200U in one axilla and placebo into the other. Two weeks later patients were injected with 100U Dysport in the axilla previously injected with placebo.
At the primary end point i.e. two weeks after treatment with Dysport, efficacy was measured as PCF (Proportional Change Function of sweat production on gravimetric analysis mg/min) relative to baseline. The results are shown as follows: (See Table 8.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn the same study, absolute sweat production was a secondary endpoint: 200U Dysport treatment resulted in an average absolute sweat production decrease from 165 ± 112 mg/min to 24 ± 27 mg/min 2 weeks after injection, and 86.2% of patients achieved an absolute sweat rate of less than 50 mg/min. The 100U treatment resulted in an average absolute sweat production decrease from 143 ± 111 mg/min to 31 ± 48 mg/min 2 weeks after injection, and 83.4% of patients achieved an absolute sweat rate of less than 50 mg/min. The placebo treatment resulted in an average absolute sweat production decrease from 173 ± 131 mg/min to 143 ± 111 mg/min 2 weeks after injection, and 3.9% of patients achieved an absolute sweat rate of less than 50 mg/min.
Efficacy was observed for up to 48 weeks. Subsequent injections under a follow up open label study showed a similar decrease in sweating though there was some evidence that duration of effect may persist for longer in subsequent treatment cycles.
Pharmacokinetics: Pharmacokinetic studies with botulinum toxin pose problems in animals because of the high potency, the minute doses involved, the large molecular weight of the compound and the difficulty of labelling toxin to produce sufficiently high specific activity. Studies using I125 labelled toxin have shown that the receptor binding is specific and saturable, and the high density of toxin receptors is a contributory factor to the high potency. Dose and time responses in monkeys showed that at low doses there was a delay of 2 - 3 days with peak effect seen 5 - 6 days after injection. The duration of action, measured by changes of ocular alignment and muscle paralysis, varied between 2 weeks and 8 months. This pattern is also seen in man, and is attributed to the process of binding, internalisation and changes at the neuromuscular junction.
Toxicology: Pre-clinical safety data: In a chronic toxicity study performed in rats, up to 12 units/animal, there was no indication of systemic toxicity. Reproductive toxicity studies in pregnant rats and rabbits given Clostridium botulinum type A toxin-haemagglutinin complex by daily intramuscular injection, at doses of 6.6 units/kg (79 units/kg total cumulative dose) and 3.0 units/kg (42 units/kg total cumulative dose) in rats and rabbits respectively, did not result in embryo/foetal toxicity. Implantation losses at maternally toxic doses were observed at higher doses in both species. Clostridium botulinum type A toxin-haemagglutinin complex demonstrated no teratogenic activity in either rats or rabbits and no effects were observed in the pre- and postnatal study on the F1 generation in rats. Fertility of male and female rats was decreased due to reduced mating secondary to muscle paralysis at doses of 29.4 units/kg weekly in males and increased implantation loss at 20 units/kg weekly in females (see Use in Pregnancy & Lactation).
In a pivotal single dose study, juveniles showed a slight delay in sexual maturation (not observed in the repeat dose study), an effect associated with decreased body weight, but subsequent mating performance and fertility were unaffected. In a pivotal repeated dose juvenile study, rats treated weekly from the age of weaning on Postnatal Day 21 up to 13 weeks of age comparable to children of 2 years old, to young adulthood (11 administrations over 10 weeks, up to total dose of approximately 33 units/kg) do not show adverse effects on postnatal growth (including skeletal evaluation), reproductive, neurological and neurobehavioral development.
The effects in reproduction, juvenile and chronic toxicity non-clinical studies were limited to changes in injected muscles related to the mechanism of action of Clostridium botulinum type A toxin-haemagglutinin complex.
There was no ocular irritation following administration of Clostridium botulinum type A toxin-haemagglutinin complex into the eyes of rabbits.