


 Sign Out
Sign Out


Pharmacology: Pharmacodynamics: Mechanism of action: Rosuvastatin is a selective, potent and competitive inhibitor of HMG-CoA reductase, the rate-limiting enzyme that converts 3-hydroxy-3-methylglutaryl coenzyme A to mevalonate, a precursor of cholesterol. Triglycerides (TG) and cholesterol in the liver are incorporated, with apolipoprotein B (ApoB), into very low density lipoprotein (VLDL) and released into the plasma for delivery to peripheral tissues. VLDL particles are TG-rich. Cholesterol-rich low density lipoprotein (LDL) is formed from VLDL and is cleared primarily through the high affinity LDL receptor in the liver.
Rosuvastatin produces its lipid-modifying effects in two ways; it increases the number of hepatic LDL receptors on the cell-surface, enhancing uptake and catabolism of LDL and it inhibits the hepatic synthesis of VLDL, thereby reducing the total number of VLDL and LDL particles.
High density lipoprotein (HDL), which contains ApoA-I is involved, amongst other things, in transport of cholesterol from tissues back to the liver (reverse cholesterol transport).
The involvement of LDL-C in atherogenesis has been well documented. Epidemiological studies have established that high LDL-C, TG, low HDL-C and ApoA-I have been linked to a higher risk of cardiovascular disease. Intervention studies have shown the benefits on mortality and CV event rates of lowering LDL-C and TG or raising HDL-C. More recent data has linked the beneficial effects of HMG-CoA reductase inhibitors to lowering of non-HDL (i.e. all circulating cholesterol not in HDL) and ApoB or reducing the ApoB/ApoA-I ratio.
Clinical efficacy: CRESTOR reduces elevated LDL-cholesterol, total cholesterol and triglycerides and increases HDL-cholesterol. It also lowers ApoB, nonHDL-C, VLDL-C, VLDL-TG and increases ApoA-I (see Table 1).
CRESTOR also lowers the LDL-C/HDL-C, total C/HDL-C, nonHDL-C/HDL-C and ApoB/ApoA-I ratio's.
A therapeutic response to CRESTOR is obtained within 1 week of commencing therapy and 90% of maximum response is usually achieved in 2 weeks. The maximum response is usually achieved by 4 weeks and is maintained after that. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe data in Table 1 are confirmed by the broader clinical programme of over 3,500 patients given CRESTOR.
In a study of patients with heterozygous familial hypercholesterolaemia, 435 subjects were given CRESTOR from 20 mg to 80 mg in a force-titration design. All doses of CRESTOR showed a beneficial effect on lipid parameters and treatment to target goals. Following titration to 40 mg (12 weeks of treatment) LDL-C was reduced by 53%.
In a force-titration open label study, 42 patients with homozygous familial hypercholesterolaemia were evaluated for their response to CRESTOR 20-40 mg titrated at a 6 week interval. In the overall population, the mean LDL-C reduction was 22%. In the 27 patients with at least a 15% reduction by week 12 (considered to be the responder population), the mean LDL-C reduction was 26% at the 20 mg dose and 30% at the 40 mg dose. Of the 13 patients with an LDL-C of less than 15%, 3 had no response or an increase in LDL-C.
In the METEOR study, the effect of CRESTOR 40 mg on the progression of atherosclerosis was assessed by B-mode ultrasound of the carotid arteries. In this multi-center, double blind, placebo-controlled clinical trial, 984 subjects at low risk for coronary heart disease (defined as Framingham risk < 10% over ten years) and with a mean LDL-C of 154.5 mg/dL but with subclinical atherosclerosis as detected by CIMT (Carotid Intima Media Thickness) were randomized in a 5:2 ratio to treatment with either CRESTOR 40 mg or placebo for 2 years. CRESTOR significantly slowed the progression of carotid atherosclerosis compared to placebo. The difference in the rate of change in the maximum CIMT of all 12 carotid artery sites between CRESTOR-treated patients and placebo-treated patients was -0.0145 mm/year (95% CI -0.0196, -0.0093; p<0.0001). The change from baseline for the CRESTOR group was -0.0014 mm/year (95% CI -0.0041, 0.0014), but was not significantly different from zero (p=0.3224). The beneficial effects of CRESTOR were consistent across all 4 secondary CIMT endpoints. There was significant progression in the placebo group (+0.0131 mm/year; 95% CI 0.0087, 0.0174; p<0.0001). In the CRESTOR group, 52.1% of patients demonstrated an absence of disease progression (i.e. regressed) compared to 37.7% of patients in the placebo group (p=0.0002). CRESTOR 40 mg was well-tolerated and the data were consistent to the established safety profile for CRESTOR.
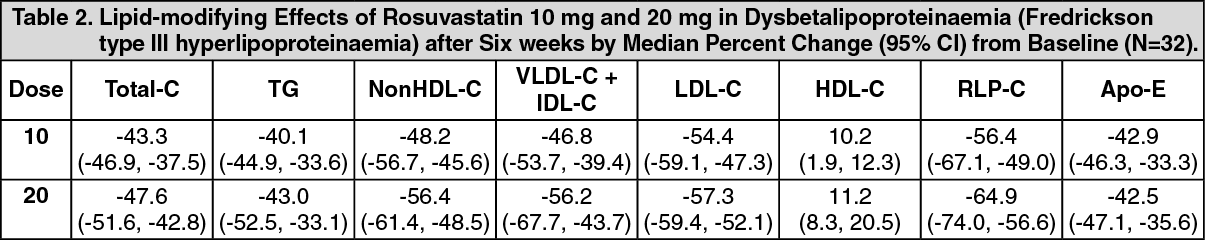
In a randomized, multicenter, double-blind crossover study, 32 patients (27 with ε2/ε2 genotype and 4 with apo E mutation [Arg145Cys]) with dysbetalipoproteinaemia (Fredrickson type III) received rosuvastatin 10 or 20 mg daily for 6 weeks. Rosuvastatin reduced non-HDL-C (primary end point) and circulating remnant lipoprotein levels. Results are shown in the table as follows. (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageCRESTOR is effective in adults with hypercholesterolaemia, with and without hypertriglyceridaemia, regardless of race, sex or age and in special populations such as diabetics or patients with familial hypercholesterolaemia.
In the Justification for the Use of Statins in Primary Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER) study, the effect of CRESTOR (rosuvastatin calcium) on the occurrence of major cardiovascular (CV) disease events was assessed in 17,802 men (≥ 50 years) and women (≥ 60 years) who had no clinically evident cardiovascular disease, LDL-C levels < 130 mg/dL (3.3 mmol/l) and hs-CRP levels ≥ 2 mg/L. The study population had an estimated baseline coronary heart disease risk of 11.6% over 10 years based on the Framingham risk criteria and included a high percentage of patients with additional risk factors such as hypertension (58%), low HDL-C levels (23%), cigarette smoking (16%) or a family history of premature CHD (12%). Study participants had a median baseline LDL-C of 108 mg/dL and hsCRP of 4.3 mg/L. Study participants were randomly assigned to placebo (n=8901) or rosuvastatin 20 mg once daily (n=8901) and were followed for a mean duration of 2 years. The JUPITER study was stopped early by the Data Safety Monitoring Board due to meeting predefined stopping rules for efficacy in rosuvastatin-treated subjects.
The primary endpoint was a composite end point consisting of the time-to-first occurrence of any of the following major CV events: CV death, nonfatal myocardial infarction, nonfatal stroke, hospitalization for unstable angina or an arterial revascularization procedure.
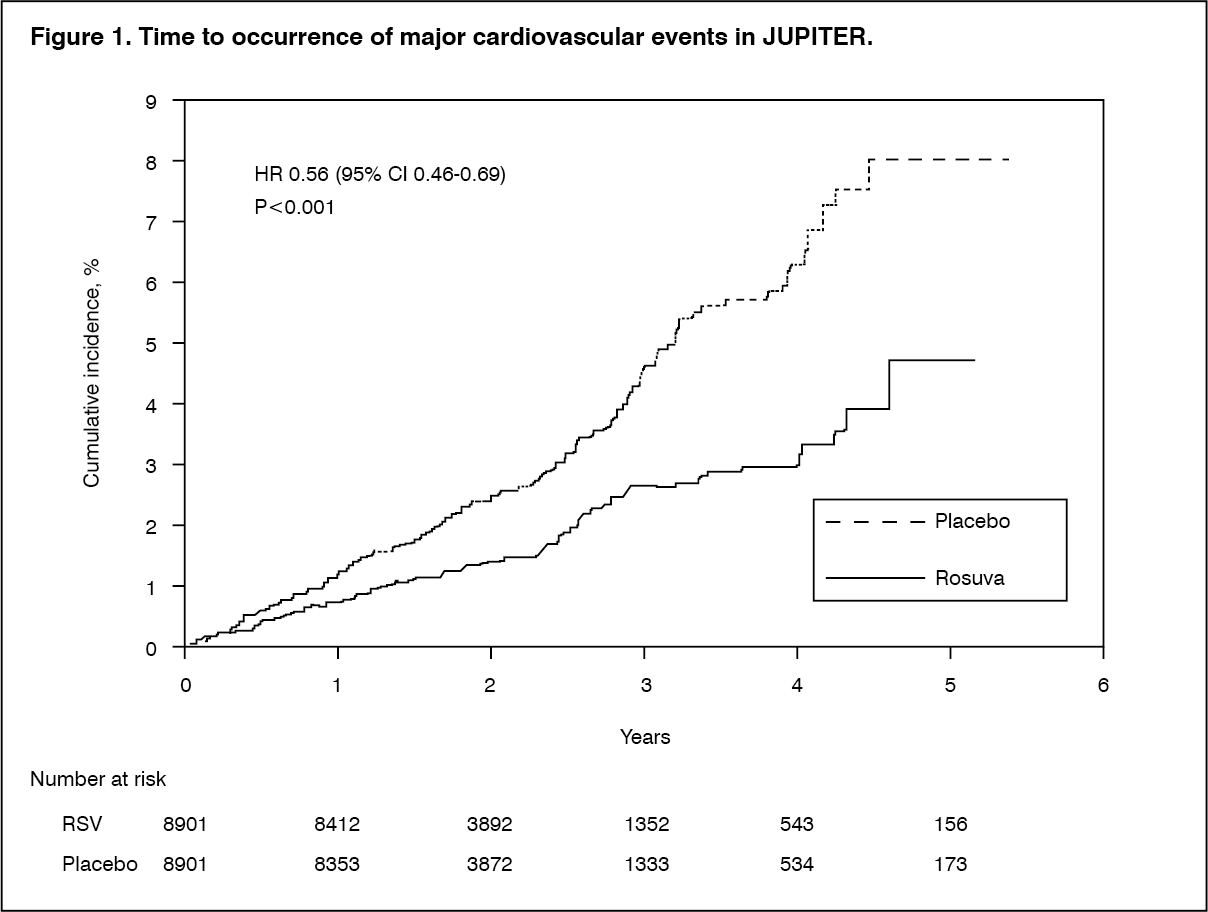
Rosuvastatin significantly reduced the risk of major CV events (252 events in the placebo group vs. 142 events in the rosuvastatin group) with a statistically significant (p<0.001) relative risk reduction of 44% and absolute risk reduction of 1.2% (see Figure 1). The risk reduction for the primary end point was consistent across the following predefined subgroups: age, sex, race, smoking status, family history of premature CHD, body mass index, LDL-C, HDL-C or hsCRP levels. (See Figure 1.)
 Click on icon to see table/diagram/image
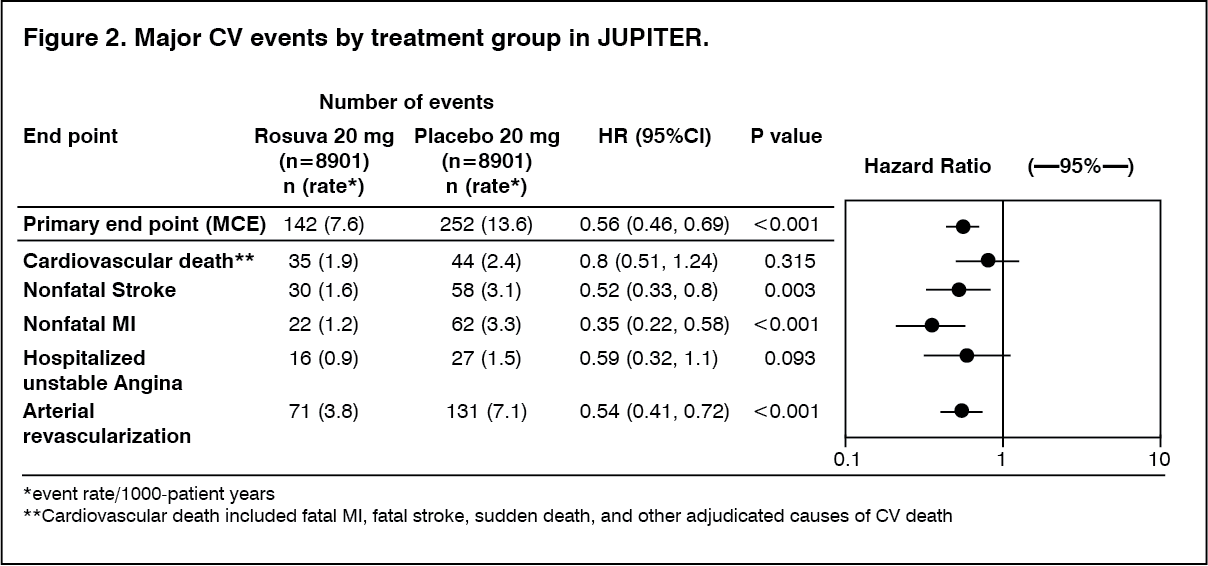
Click on icon to see table/diagram/imageThe individual components of the primary end point are presented in Figure 2. Rosuvastatin significantly reduced the risk of nonfatal myocardial infarction, nonfatal stroke, and arterial revascularization procedures. There were no significant treatment differences between the rosuvastatin and placebo groups for death due to cardiovascular causes or hospitalizations for unstable angina.
Rosuvastatin significantly reduced the risk of myocardial infarction (6 fatal events and 62 nonfatal events in placebo-treated subjects vs. 9 fatal events and 22 nonfatal events in rosuvastatin-treated subjects) and the risk of stroke (6 fatal events and 58 nonfatal events in placebo-treated subjects vs. 3 fatal events and 30 nonfatal events in rosuvastatin-treated subjects).
In JUPITER, there was a statistically significant increase in the frequency of diabetes mellitus reported by investigators; 2.8% of patients in the rosuvastatin group and 2.3% of patients in the placebo group (HR: 1.27, 95% CI: 1.05-1.53, p=0.015). The difference between treatment groups (rosuvastatin versus placebo) in mean HbA1c change from baseline was approximately 0.1%. The cardiovascular and mortality benefits of rosuvastatin therapy exceeded the diabetes hazard in the trial population as a whole (see Precautions and Adverse Reactions).
In a post-hoc subgroup analysis of JUPITER subjects (n=1405; rosuvastatin=725, placebo=680) with a hsCRP ≥ 2 mg/L and no other traditional risk factors (smoking, BP ≥ 140/90 or taking antihypertensives, low HDL-C) other than age, after adjustment for high HDL-C, there was no significant treatment benefit with rosuvastatin treatment. (See Figure 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAt one year, rosuvastatin increased HDL-C and reduced LDL-C, hsCRP, total cholesterol and serum triglyceride levels (p<0.001 for all versus placebo).
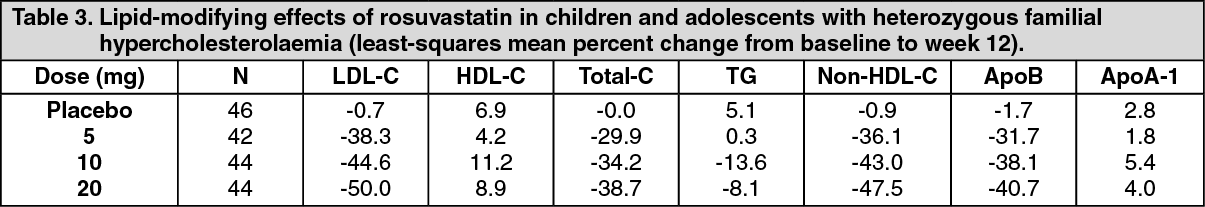
Children and Adolescents with Hypercholesterolaemia: In a double blind, randomized, multi-centre, placebo-controlled, 12-week study (n=176, 97 male and 79 female) followed by a 40-week (n=173, 96 male and 77 female), open label, rosuvastatin dose titration phase, 10-17 years of age (Tanner stage II-V, females at least 1 year post-menarche) with heterozygous familial hypercholesterolaemia received rosuvastatin 5, 10 or 20 mg or placebo daily for 12 weeks and then all received rosuvastatin daily for 40 weeks. At study entry, approximately 30% of the patients were 10-13 years and approximately 17%, 18%, 40%, and 25% were Tanner stage II, III, IV, and V respectively.
Rosuvastatin reduced LDL-C (primary end point), total cholesterol and ApoB levels. Results are shown in Table 3 as follows. (See Table 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAt the end of the 40 week, open label, titration to goal, dosing up to a maximum of 20 mg once daily, 70 of 173 patients (40.5%) had achieved the LDL-C goal of less than 110 mg/dL (2.8 mmol/L).
After 52 weeks' of study treatment, no effect on growth or sexual maturation was detected (see Precautions).
Pharmacokinetics: CRESTOR is administered orally in the active form with peak plasma levels occurring 5 hours after dosing. Exposure increases linearly over the dose range. The half life is 19 hours and does not increase with increasing dose. Absolute bioavailability is 20%. There is minimal accumulation on repeated once daily dosing.
Rosuvastatin undergoes first pass extraction in the liver which is the primary site of cholesterol synthesis and LDL-C clearance.
Rosuvastatin is approximately 90% bound to plasma proteins, mostly albumin. The parent compound, accounts for greater than 90% of the circulating active HMG CoA reductase inhibitor activity.
Rosuvastatin undergoes limited metabolism (approximately 10%), mainly to the N-desmethyl form, and 90% is eliminated as unchanged drug in the faeces with the remainder being excreted in the urine.
Special populations: Age and sex: There was no clinically relevant effect of age or sex on the pharmacokinetics of rosuvastatin in adults. The pharmacokinetics of rosuvastatin in children and adolescents with heterozygous familial hypercholesterolaemia was similar to that of adult volunteers.
Renal insufficiency: In a study in subjects with varying degrees of renal impairment, mild to moderate renal disease had little influence on plasma concentrations of rosuvastatin. However, subjects with severe impairment (CrCl < 30 ml/min) had a 3-fold increase in plasma concentration compared to healthy volunteers. Steady-state plasma concentrations of rosuvastatin in subjects undergoing haemodialysis were approximately 50% greater compared to healthy volunteers.
Hepatic insufficiency: In a study in 12 subjects with mild to moderate hepatic impairment there was evidence of increased exposure to rosuvastatin in the 2 subjects with the higher Child-Pugh scores (8 and 9). In these subjects systemic exposure was increased by at least 2-fold compared to subjects with lower Child-Pugh scores. There is no experience in subjects with Child-Pugh scores above 9.
Race: Pharmacokinetic studies show an approximate 2-fold elevation in median AUC in Asian subjects including subjects of Japanese, Chinese, Malay and Indian ancestry compared with Caucasians. A population pharmacokinetic analysis revealed no clinically relevant differences in pharmacokinetics among Caucasian, Hispanic and Black or Afro-Caribbean groups.
Genetic polymorphisms: Disposition of HMG-CoA reductase inhibitors, including rosuvastatin, involves OATP1B1 and BCRP transporter proteins. In patients with SLCO1B1 (OATP1B1) and/or ABCG2 (BCRP) genetic polymorphisms there is a risk of increased rosuvastatin exposure. Individual polymorphisms of SLCO1B1 c.521CC and ABCG2 c.421AA are associated with an approximate 1.6-fold higher rosuvastatin exposure (AUC) or 2.4-fold higher exposure, respectively, compared to the SLCO1B1 c.521TT or ABCG2 c.421CC genotypes.
Toxicology: Preclinical safety data: Preclinical data reveal no special hazards for humans based on conventional studies of safety pharmacology, repeat-dose toxicity, genotoxicity, carcinogenic potential, and reproductive toxicity.