Because clinical trials are conducted under very specific conditions the adverse drug reaction rates observed in the clinical trials may not reflect the rates observed in practice and should not be compared to the rates in the clinical trials of another drug. Adverse drug reaction information from clinical trials is useful for identifying drug-related adverse events and for approximating rates.
Clinical Studies: In clinical studies, alendronate was generally well tolerated. In studies of up to five years in duration, side effects, which usually were mild, generally did not require discontinuation of therapy.
Alendronate has been evaluated for safety in clinical studies in approximately 7200 postmenopausal women.
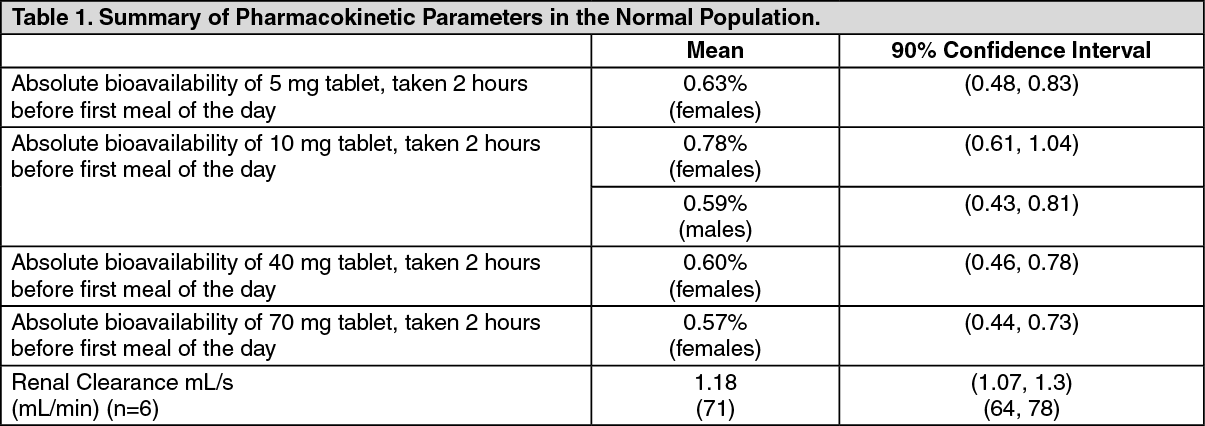
Treatment of Osteoporosis: Postmenopausal Women: In two, three-year, placebo-controlled, double-blind, multicenter studies (United States and Multinational) of virtually identical design, with a total of 994 postmenopausal women, the overall safety profiles of alendronate 10 mg/day and placebo were similar. Discontinuation of therapy due to any clinical adverse experience occurred in 4.1% of 196 patients treated with alendronate 10 mg/day and 6.0% of 397 patients treated with placebo.
Adverse experiences considered by the investigators as possibly, probably, or definitely drug-related in >1% of patients treated with either alendronate 10 mg/day or placebo are presented in the following table. (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Less Common Clinical Adverse Reactions (<1%): Rarely, rash and erythema have occurred.
One patient treated with alendronate (10 mg/day), who had a history of peptic ulcer disease and gastrectomy and who was taking concomitant acetylsalicylic acid (ASA) developed an anastomotic ulcer with mild hemorrhage, which was considered drug-related. ASA and alendronate were discontinued and the patient recovered.
In the two-year extension (treatment years 4 and 5) of the previously mentioned studies, the overall safety profile of alendronate 10 mg/day was similar to that observed during the three-year placebo-controlled period. Additionally, the proportion of patients who discontinued alendronate 10 mg/day due to any clinical adverse experience was similar to that during the first three years of the study.
In the Fracture Intervention Trial, discontinuation of therapy due to any clinical adverse experience occurred in 9.1% of 3236 patients treated with alendronate 5 mg/day for two years and 10 mg/day for either one or two additional years and 10.1% of 3223 patients treated with placebo.
Discontinuations due to upper gastrointestinal adverse experiences were: alendronate, 3.2%; placebo, 2.7%. The overall adverse experience profile was similar to that seen in other studies with alendronate 5 or 10 mg/day.
In a one-year, double-blind multicenter study, the overall safety and tolerability profiles of alendronate 70 mg once weekly and alendronate 10 mg daily were similar. The adverse experiences considered by the investigators as possibly, probably, or definitely drug-related in >1% of patients in either treatment group are presented in the following table: See Table 3.
Click on icon to see table/diagram/image
Men: In two, placebo-controlled, double-blind, multicenter studies in men (a two-year study of alendronate 10 mg/day (n=146) and a one-year study of alendronate 70 mg once weekly (n=109)), the safety profile of alendronate was generally similar to that seen in postmenopausal women. The rates of discontinuation of therapy due to any clinical adverse experience were 2.7% for alendronate 10 mg/day vs 10.5% for placebo and 6.4% for alendronate 70 mg once weekly vs 8.6% for placebo.
Other studies in men and women: In a ten-week endoscopy study in men and women (n=277; mean age: 55) no difference was seen in upper gastrointestinal tract lesions between alendronate 70 mg once weekly and placebo.
In an additional one-year study in men and women (n=335; mean age: 50) the overall safety and tolerability profiles of alendronate 70 mg once weekly were similar to that of placebo and no difference was seen between men and women.
Prevention of Osteoporosis in Postmenopausal Women: The safety of alendronate 5 mg/day in postmenopausal women 40-60 years of age has been evaluated in three double-blind, placebo-controlled studies involving over 1400 patients randomized to receive alendronate for either two or three years. In these studies the overall safety profiles of alendronate 5 mg/day and placebo were similar. Discontinuation of therapy due to any clinical adverse experience occurred in 7.5% of 642 patients treated with alendronate 5 mg/day and 5.7% of 648 patients treated with placebo. Adverse experiences reported by the investigators as possibly, probably or definitely drug related in >1% of patients treated with either alendronate 5 mg/day or placebo are presented in the following table: See Table 4.
Click on icon to see table/diagram/image
Concomitant Use with Estrogen/Hormone Replacement Therapy: In two studies (of one and two years duration) of postmenopausal osteoporotic women (total: n=853), the safety and tolerability profile of combined treatment with alendronate 10 mg once daily and estrogen ± progestin (n=354) was consistent with those of the individual treatments.
Treatment and Prevention of Glucocorticoid-Induced Osteoporosis: In two, one-year, placebo-controlled, double-blind, multicenter studies in patients receiving glucocorticoid treatment, the overall safety and tolerability profiles of alendronate 5 or 10 mg/day were generally similar to that of placebo. Adverse experiences reported by the investigators as possibly, probably or definitely drug related in >1% of patients treated with either alendronate 5 or 10 mg/day or placebo are presented in the following table: See Table 5.
Click on icon to see table/diagram/image
The overall safety and tolerability profile in the glucocorticoid-induced osteoporosis population that continued therapy for the second year of the studies was consistent with that observed in the first year.
Paget's Disease of Bone: In clinical studies (Paget's disease and osteoporosis), adverse experiences reported in 175 patients taking alendronate 40 mg/day for 3-12 months were similar to those in postmenopausal women treated with alendronate 10 mg/day. However, there was an apparent increased incidence of upper gastrointestinal adverse experiences in patients taking alendronate 40 mg/day (17.7% alendronate vs 10.2% placebo). Isolated cases of esophagitis and gastritis resulted in discontinuation of treatment.
Additionally, musculoskeletal pain (bone, muscle or joint), which has been described in patients with Paget's disease treated with other bisphosphonates, was reported by the investigators as possibly, probably, or definitely drug-related in approximately 6% of patients treated with alendronate 40 mg/day versus approximately 1% of patients treated with placebo, but rarely resulted in discontinuation of therapy. Discontinuation of therapy due to any clinical adverse experience occurred in 6.4% of patients with Paget's disease treated with alendronate 40 mg/day and 2.4% of patients treated with placebo.
Post-Marketing Experience: The following adverse reactions have been reported in post-marketing use:
Body as a Whole: Hypersensitivity reactions including urticaria and rarely angioedema. As with other bisphosphonates, transient symptoms as in an acute-phase response (myalgia, malaise and rarely, fever) have been reported with alendronate, typically in association with initiation of treatment.
Rarely, symptomatic hypocalcemia has occurred, both in association with predisposing conditions and in patients without known predisposing conditions.
Gastrointestinal: Esophagitis, esophageal erosions, esophageal ulcers, rarely esophageal stricture or perforation, and oropharyngeal ulceration. Some of these have been serious and required hospitalization. Rarely, gastric or duodenal ulcers, some severe and with complications, although a causal relationship has not been established (see Dosage & Administration and Precautions).
Skin: Rash (occasionally with photosensitivity), pruritus, rarely severe skin reactions, including Stevens-Johnson syndrome and toxic epidermal necrolysis.
Special Senses: Rarely uveitis, rarely scleritis.
Osteonecrosis of the jaw: Osteonecrosis of the jaw has been reported in patients receiving treatment regimens including bisphosphonates (see PRECAUTIONS). Osteonecrosis of the jaws has other well documented multiple risk factors. It is not possible to determine if these events are related to alendronate or other bisphosphonates, to concomitant drugs or other therapies (e.g., chemotherapy, head and neck radiotherapy, corticosteroid), to patient's underlying disease or to other co-morbid risk factors (e.g., anemia, infection, preexisting oral disease).
Laboratory Tests: In double-blind, multicenter, controlled studies, asymptomatic, mild, and transient decreases in serum calcium and phosphate were observed in approximately 18 and 10%, respectively, of patients taking alendronate versus approximately 12 and 3% of those taking placebo. However, the incidences of decreases in serum calcium to <8.0 mg/dL (2.0 mM) and serum phosphate to <2.0 mg P1/dL (0.65 mM) were similar in both treatment groups.
In a small, open label study, at higher doses (80 mg/day) some patients had elevated transaminases. However, this was not observed at 40 mg/day. No clinically significant toxicity was associated with these laboratory abnormalities.
Rare cases of leukemia have been reported following therapy with other bisphosphonates. Any causal relationship to either the treatment or to the patients' underlying disease has not been established.
1P: Elemental phosphorus.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image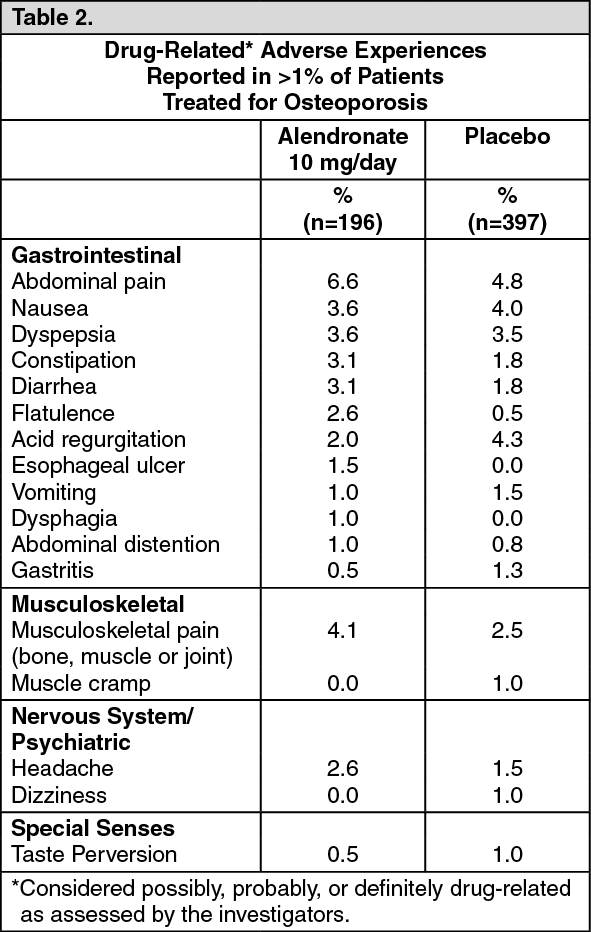 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image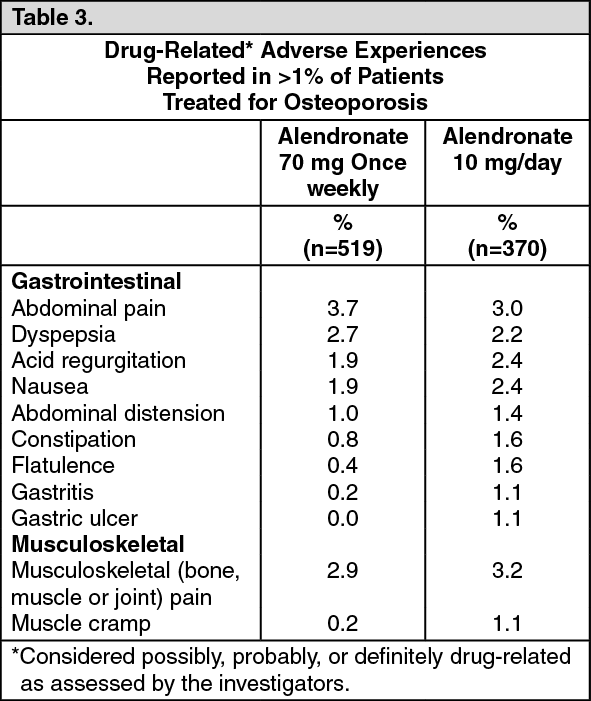 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
