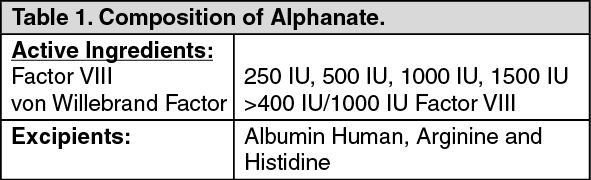
Pharmacology: Pharmacodynamics: Mechanism of Action: Antihemophilic Factor/von Willebrand Factor Complex (Human) (Factor VIII) and von Willebrand Factor (VWF) are constituents of normal plasma and are required for clotting. The administration of Alphanate temporarily increases the plasma level of Factor VIII, thus minimizing the hazard of hemorrhage. Factor VIII is an essential cofactor in activation of Factor X leading to formation of thrombin and fibrin. VWF promotes platelet aggregation and platelet adhesion on damaged vascular endothelium; it also serves as a stabilizing carrier protein for the procoagulant protein Factor VIII.
Clinical Studies: Prophylaxis for Elective Surgery: Thirty seven subjects with VWD (6 Type 1, 16 Type 2A, 3 Type 2B, 12 Type 3) underwent 59 surgical procedures that included 20 dental, 7 orthopedic, 8 gastrointestinal, 6 gastrointestinal (diagnostic), 9 vascular, 3 gynecologic, 2 genitourinary, 2 dermatologic and 2 head and neck procedures administering A-SD or A-SD/HT (21 subjects were administered A-SD and 18 were administered A-SD/HT, 2 received both products) for bleeding prophylaxis (see Table 3). Prior to each surgical procedure, the investigators provided an estimation of the expected blood loss during surgery for a normal person of the same sex and of similar stature and age as the subject undergoing the same type of surgical procedure. An initial preoperative infusion of 60 VWF:RCo IU/kg (75 VWF:RCo IU/kg for patients less than 18 years of age), was administered one hour preoperatively. A sample was obtained 15 minutes after the initial infusion for the determination of the plasma FVIII:C level. The level had to equal or exceed 100% of normal for an operation to proceed. No cryoprecipitate or alternative FVIII product was administered during these surgical procedures. Platelets were required in only two subjects. Intra-operative infusions of A-SD and A-SD/HT at 60 VWF:RCo IU/kg (75 VWF:RCo IU/kg for patients less than 18 years of age) was administered according to the judgment of the investigator. (See Table 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Postoperative infusions at doses of 40 to 60 VWF:RCo IU/kg (50 to 75 VWF:RCo IU/kg for pediatric patients) was administered at 8- to 12-hour intervals until healing had occurred. After achieving primary hemostasis, for maintenance of secondary hemostasis the dose was reduced after the third postoperative day.
Overall, in 55 surgical procedures undertaken with a prolonged BT pre-infusion, the BT at 30 minutes post-infusion was fully corrected in 18 (32.7%) cases, partially corrected in 24 (43.6%) cases, demonstrated no correction in 12 (21.8%) cases, and was not done in one case (1.8%).
The mean blood loss was lower than predicted prospectively. Bleeding exceeding the predicted value did not correlate with correction of the BT. Three patients had bleeding which exceeded by more than 50 mL the amount predicted prospectively. Among the latter subjects, the BT 30 minutes post-infusion was normal in one and only slightly lengthened in two cases.
Surgical infusion summary data are included in Table 4. (See Table 4.)
Click on icon to see table/diagram/image
Additionally, the surgeries were categorized as major, minor or invasive procedures according to definitions used in the study. The outcome of each surgery was evaluated according to a clinical rating scale (excellent, good, poor or none) and was considered successful if the outcome was excellent or good. These outcomes are presented in Table 5. (See Table 5.)
Click on icon to see table/diagram/image
The study results were also evaluated independently by two referees with clinical experience in this field in the same way (surgery categorization and outcome of each surgery according to a clinical rating scale).
The results for the effect of treatment on surgical prophylaxis (Referee Evaluation) per treated subject are summarized in Table 8. There is a high level of agreement between the referee evaluations and the analyzed outcome data, with a decrease of only a single success (21/24 vs. 22/24). (See Table 6.)
Click on icon to see table/diagram/image
A retrospective study was performed to assess the efficacy of Alphanate (A-SD/HT) as replacement therapy in preventing excessive bleeding in subjects with congenital VWD undergoing surgical or invasive procedures, for whom DDAVP was ineffective or inadequate. The study was performed between September 2004 and December 2005, and 61 surgeries/procedures (in 39 subjects) were evaluated.
Of the 39 subjects, 18 had Type 1 VWD (46.2%); 12 subjects (30.8%) had Type 2 VWD, and 9 subjects (23.1%) had Type 3 VWD. The median age for subjects overall was 40 years; approximately one-half of the subjects overall were male.
The primary efficacy variable was the overall treatment outcome for each surgical or invasive procedure, as rated by the investigator using a 4-point verbal rating scale (VRS): "excellent," "good," "poor," or "none." The categorization of the replacement treatment outcome according to the proposed scale was based upon the investigator's clinical experience.
The secondary efficacy variables were: Daily (Day 0 and Day 1) treatment outcome for each surgical or invasive procedure, rated by the investigator using the same 4-point VRS used for the primary efficacy variable. Day 0 was the day of surgery, and Day 1 was the day following surgery.
Overall treatment outcome for each surgical or invasive procedure, rated by an independent referee committee using the same 4-point VRS used for the primary efficacy variable.
In addition, an independent referee committee was convened to evaluate the efficacy outcomes. The committee was composed of 2 physicians with demonstrated clinical expertise treating subjects with similar medical characteristics to those of the study population. The committee was blinded to the investigator ratings; and each referee evaluated the outcomes independent of one another.
More than 90% received an investigator and referee's overall and daily rating of "effective" ("excellent" or "good"). The results of the primary efficacy analysis are in Table 7. (See Table 7.)
Click on icon to see table/diagram/image
The results of the analysis of daily investigator ratings are in Table 8. (See Table 8.)
Click on icon to see table/diagram/image
The results of the analysis of overall referee ratings are in Table 9. (See Table 9.)
Click on icon to see table/diagram/image
The overall investigator ratings are summarized by type of VWD in Table 10. (See Table 10.)
Click on icon to see table/diagram/image
The majority of ratings were "excellent" (≥81.3% in each VWD type). Only 2 procedures in 1 subject with Type 3 VWD received an overall efficacy rating of "none," and 1 procedure in 1 subject with Type 2 VWD received an overall efficacy rating of "poor."
The total dose of Alphanate received over the entire perioperative period of the retrospective study is summarized in Table 11. (See Table 11.)
Click on icon to see table/diagram/image
Pharmacokinetics: Pharmacokinetics in Hemophilia A: Following the administration of Alphanate during clinical trials, the mean
in vivo half-life of Factor VIII observed in 12 adult subjects with severe hemophilia A was 17.9 ± 9.6 hours. In this same study, the
in vivo recovery was 96.7 ± 14.5% at 10 minutes postinfusion. Recovery at 10 minutes post-infusion was also determined as 2.4 ± 0.4 IU FVIII rise/dL plasma per IU FVIII infused/kg body weight.
Pharmacokinetics in von Willebrand Disease (VWD): A pharmacokinetic crossover study was conducted in 14 non-bleeding subjects with VWD (1 type 1, 2 type 2A, and 11 type 3) comparing the pharmacokinetics of Alphanate SD/HT (A-SD/HT) and an earlier formulation, Alphanate SD (A-SD), which was treated with solvent-detergent but was not heat-treated. Subjects received, in random order at least seven days apart, a single intravenous dose each of A-SD and A-SD/HT, 60 VWF:RCo IU/kg (75 VWF:RCo IU/kg in subjects younger than 18 years of age). Pharmacokinetic parameters were similar for the two preparations and indicated that they were biochemically equivalent. Pharmacokinetic analysis of A-SD/HT in the 14 subjects revealed the following results: the median plasma levels of VWF:RCo rose from 0.17 IU/dL [mean, 0.2 ± 0.08 IU/dL; range: 0.1 to 0.5 IU/dL] at baseline to 3.43 IU/dL [mean, 3.5 ± 1.47 IU/dL; range: 1.5 to 5.9 IU/dL] 15 minutes post-infusion; median plasma levels of FVIII:C rose from 0.08 IU/dL [mean, 0.2 ± 0.34 IU/dL; range: 0.0 to 1.2 IU/dL] to 2.14 IU/dL [mean, 2.4 ± 0.72 IU/dL; range: 1.4 to 3.9 IU/dL]. The median bleeding time (BT) prior to infusion was 30 minutes (mean, 28.8 ± 4.41 minutes; range: 13.5 to 30 minutes), which shortened to 10.38 minutes (mean, 10.4 ± 3.20 minutes; range: 6 to 16 minutes) 1 hour post-infusion.
Following infusion of A-SD/HT, the median half-lives for VWF:RCo, FVIII:C and VWF:Ag were 6.91 hours (mean, 7.46 ± 3.20 hours, range, 3.68 to 16.22 hours), 20.87 hours (mean, 21.52 ± 7.21 hours; range: 7.19 to 32.20 hours), and 12.66 hours (mean, 13.03 ± 2.12 hours: range: 10.34 to 17.45 hours), respectively. The median incremental
in vivo recoveries of VWF:RCo and FVIII:C were 3.12 (IU/dL)/(IU/kg) [mean, 3.29 ± 1.46 (IU/dL)/(IU/kg); range: 1.3 to 5.7 (IU/dL)/(IU/kg)] for VWF:RCo and 1.94 (IU/dL)/(IU/kg) [mean, 2.14 ± 0.58 (IU/dL)/(IU/kg); range: 1.3 to 3.3 (IU/dL)/(IU/kg)] for FVIII:C.
Following infusion of both A-SD and A-SD/HT, an increase in the size of VWF multimers was seen and persisted for at least 24 hours. The shortening of the BT was transient, lasting less than 6 hours following treatment and did not correlate with the presence of large and intermediate size VWF multimers.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image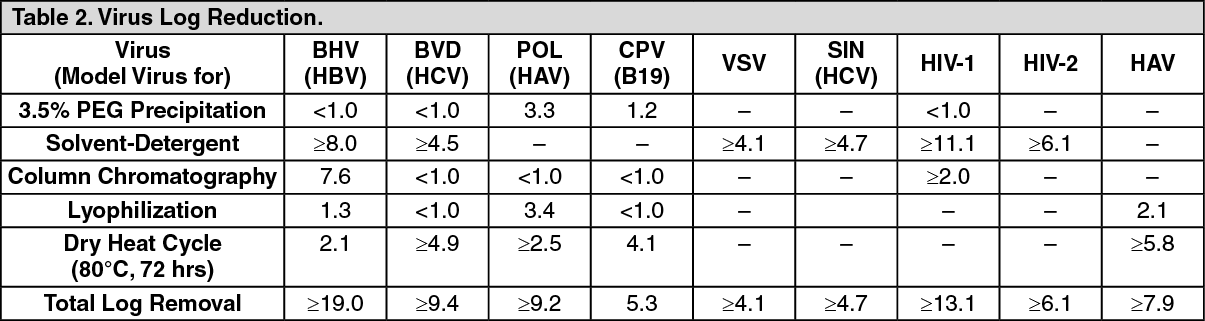 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image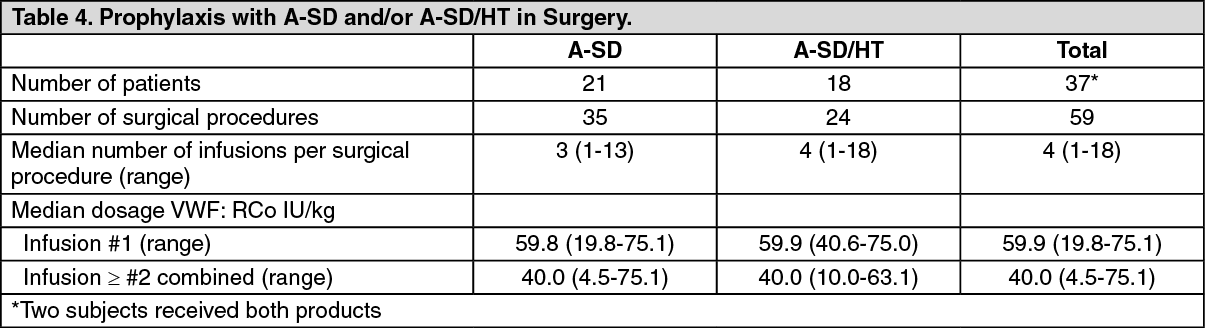 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image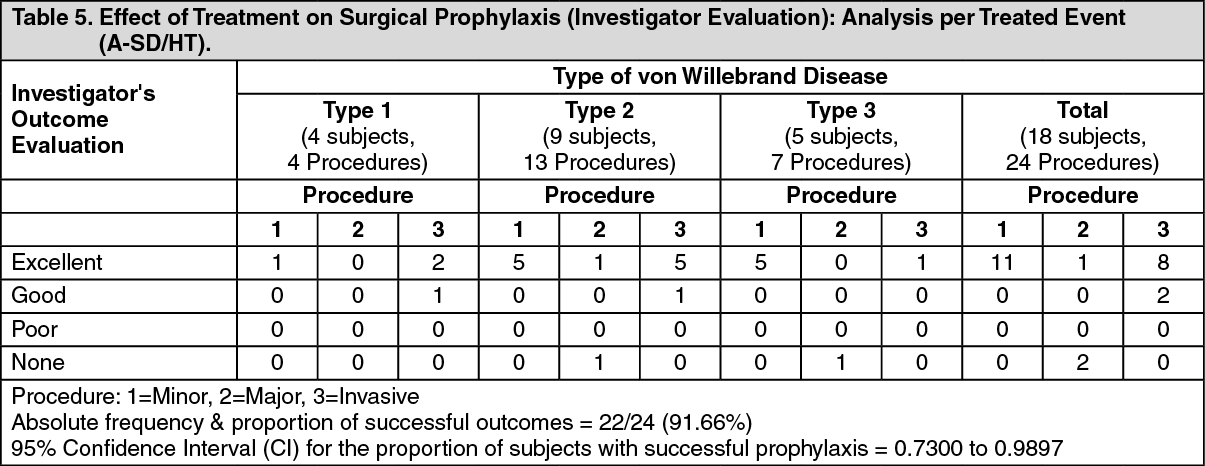 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image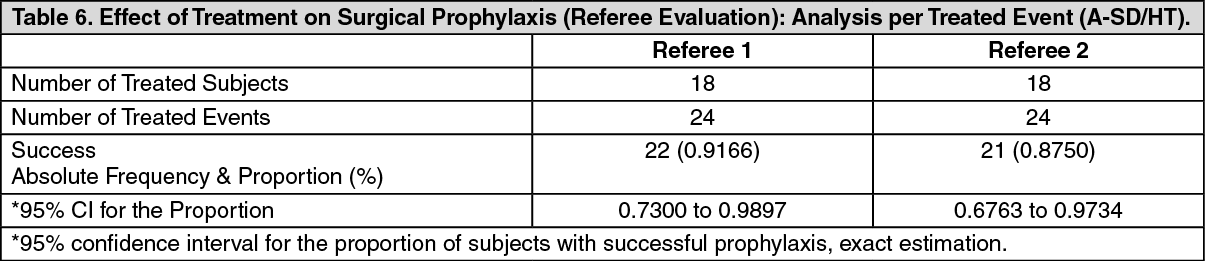 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image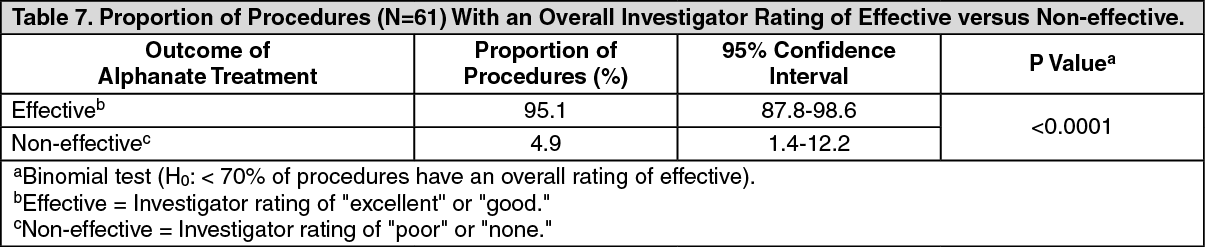 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image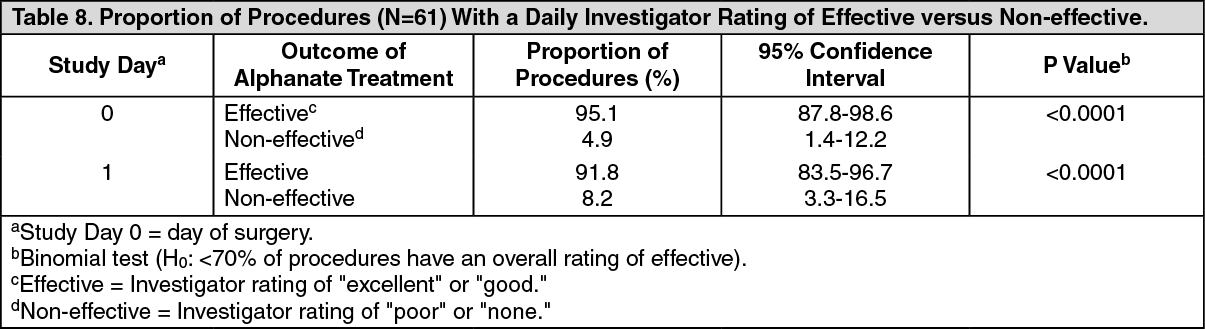 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image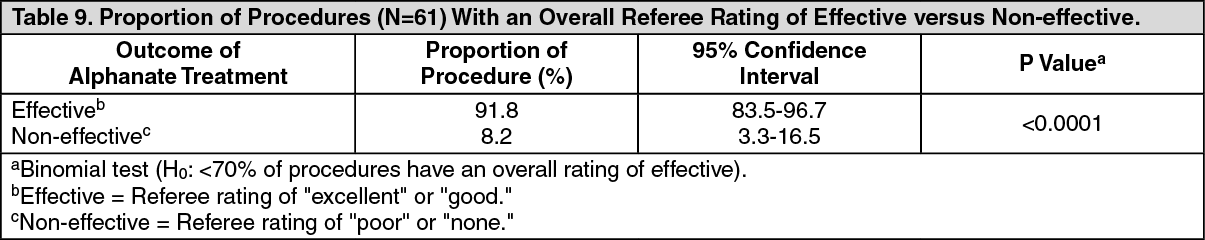 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image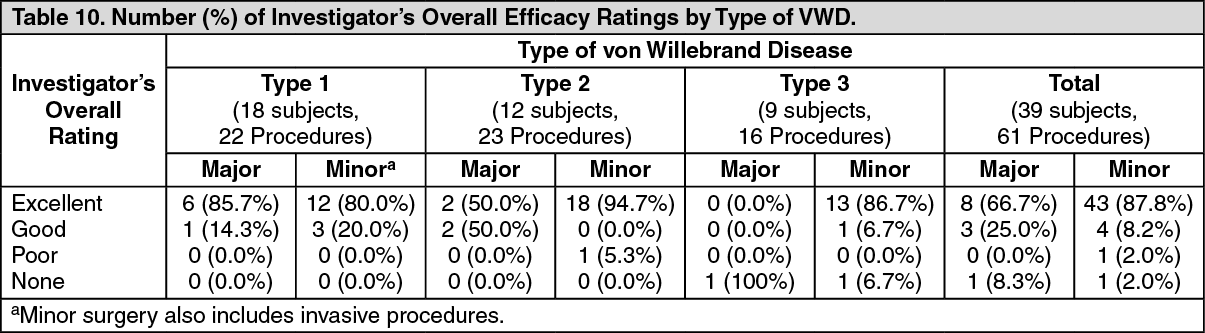 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image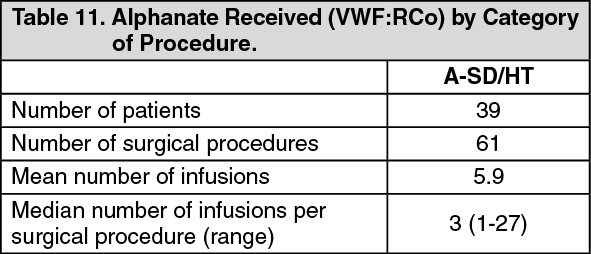 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
