Pharmacotherapeutic group: antiinflammatory and antirheumatic products, non-steroids, coxibs.
ATC code: M01AH01.
Pharmacology: Pharmacodynamics: Mechanism of action: The mechanism of action of celecoxib is via inhibition of prostaglandin synthesis primarily by inhibition of COX-2. At therapeutic concentrations in humans celecoxib does not inhibit cyclooxygenase-1 (COX-1). COX-2 is induced in response to inflammatory stimuli. This leads to the synthesis and accumulation of inflammatory prostanoids, in particular prostaglandin E2, causing inflammation, oedema and pain. Celecoxib acts as an anti-inflammatory, analgesic, and antipyretic agent in animal models by blocking the production of inflammatory prostanoids via COX-2 inhibition.
In vivo and
ex vivo studies show that celecoxib has a very low affinity for the constitutively expressed COX-1 enzyme. Consequently at therapeutic doses celecoxib has no effect on prostanoids synthesized by activation of COX-1 thereby not interfering with normal COX-1 related physiological processes in tissues, particularly the stomach, intestine and platelets.
Clinical Studies: Osteoarthritis (OA): Celecoxib has demonstrated significant reduction in joint pain compared to placebo. Celecoxib was evaluated for treatment of the signs and the symptoms of OA of the knee and hip in approximately 4,200 patients in placebo- and active-controlled clinical trials of up to 12 weeks duration. In patients with OA, treatment with celecoxib 100 mg twice daily or 200 mg once daily resulted in improvement in WOMAC (Western Ontario and McMaster Universities) osteoarthritis index, a composite of pain, stiffness, and functional measures in OA. In three 12-week studies of pain accompanying OA flare, celecoxib doses of 100 mg twice daily or 200 mg twice daily provided significant reduction of pain within 24 to 48 hours of initiation of dosing. At doses of 100 mg twice daily or 200 mg twice daily the efficacy of celecoxib was shown to be similar to that of naproxen 500 mg twice daily. Doses of 200 mg twice daily provided no additional benefit above that seen with 100 mg twice daily. A total daily dose of 200 mg has been shown to be equally effective whether administered as 100 mg twice daily or 200 mg once daily.
Rheumatoid Arthritis (RA): Celecoxib has demonstrated significant reduction in joint tenderness/pain and joint swelling compared to placebo. Celecoxib was evaluated for treatment of the signs and symptoms of RA in approximately 2100 patients in placebo- and active-controlled clinical trials of up to 24 weeks in duration. Celecoxib was shown to be superior to placebo in these studies, using the American College of Rheumatology 20 (ACR20) Responder Index, a composite of clinical, laboratory, and functional measures in RA. Celecoxib doses of 100 mg twice daily and 200 mg twice daily were similar in efficacy and both were comparable to naproxen 500 mg twice daily.
Although celecoxib 100 mg twice daily and 200 mg twice daily provided similar overall efficacy, some patients derived additional benefit from the 200 mg twice daily dose. Doses of 400 mg twice daily provided no additional benefit above that seen with 100 mg to 200 mg twice daily.
Analgesia, including Primary Dysmenorrhea: In acute analgesic models of post-oral surgery pain, post-orthopedic surgical pain, and primary dysmenorrhea, celecoxib relieved pain that was rated by patients as moderate to severe. Single doses of celecoxib provided pain relief within 60 minutes (see Dosage & Administration).
Ankylosing Spondylitis (AS): Celecoxib was evaluated in AS patients in two placebo- and active controlled (naproxen or ketoprofen) clinical trials of 6 and 12 weeks duration. Celecoxib at doses of 100 mg twice daily, 200 mg once daily and 400 mg once daily was shown to be statistically superior to placebo in these studies for all three co-primary efficacy measures assessing global pain intensity (Visual Analogue Scale), global disease activity (Visual Analogue Scale), and functional impairment (Bath Ankylosing Spondylitis Functional Index). In the 12-week study, there was no difference in the extent of improvement between the 200 mg and 400 mg celecoxib doses in a comparison of mean change from baseline, but there was a greater percentage of patients who responded to celecoxib 400 mg, 53%, than to celecoxib 200 mg, 44%, using the Assessment in Ankylosing Spondylitis response criteria (ASAS 20). The ASAS 20 defines response as improvement from baseline of at least 20% and an absolute improvement of at least 10 mm, on a 0 to 100 mm scale, in at least three of the four following domains: Patient Global Assessment of Disease, Patient's Global Pain Intensity, Bath Ankylosing Spondylitis Functional Index, and inflammation. The responder analysis also demonstrated no change in the responder rates beyond 6 weeks.
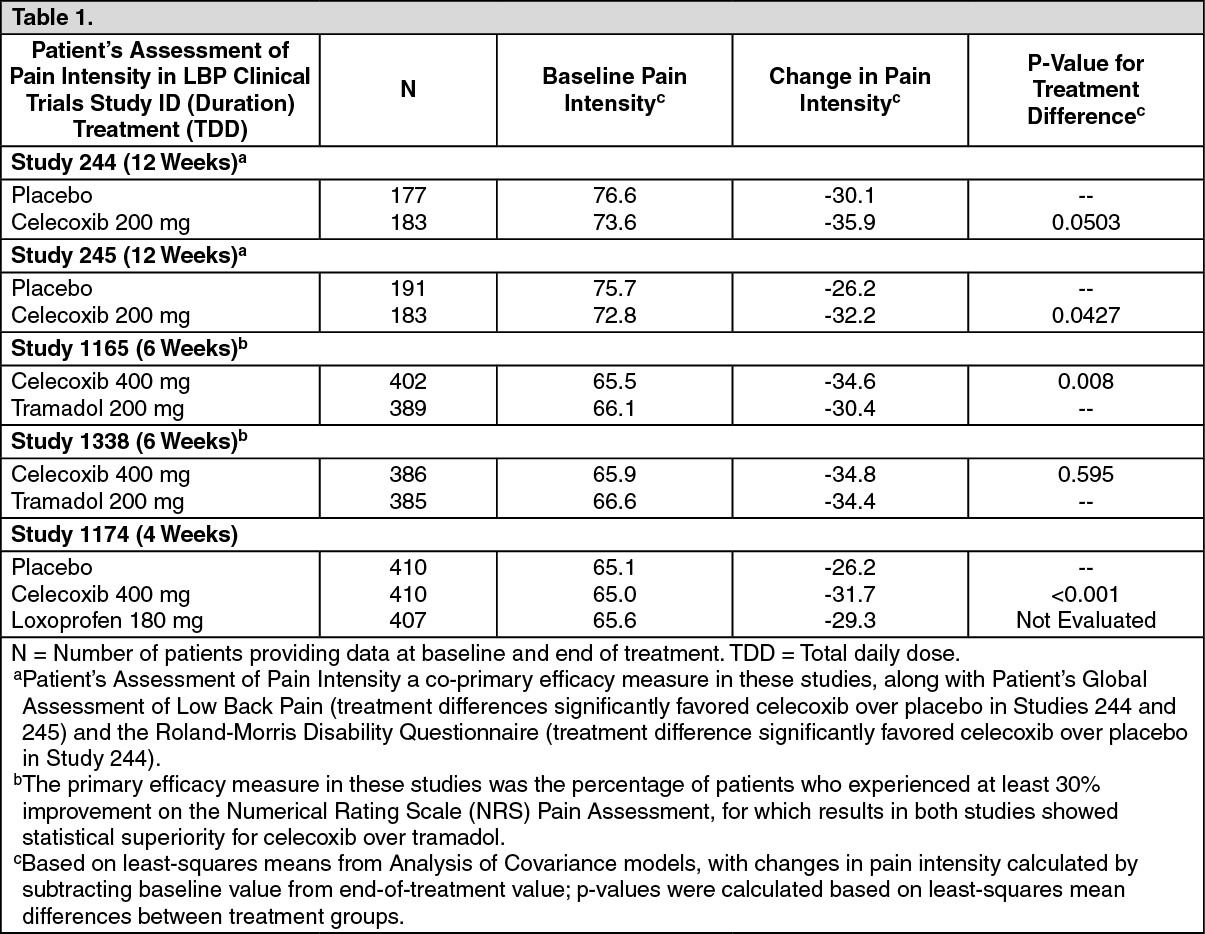
Chronic Low Back Pain (LBP): Celecoxib was used to treat patients who had pre-existing non-neuropathic LBP of duration ≥12 weeks. In the table shown as follows, efficacy results in 5 clinical trials of up to 12 weeks duration are presented using the Patient's Assessment of Pain Intensity (100 mm visual analog scale) from baseline to end of treatment: (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Further Information from Clinical Studies: Endoscopic Studies: Five randomised double-blind controlled trials have been conducted including scheduled upper gastrointestinal endoscopy in over 4,000 patients with OA and RA in which ulceration rates on celecoxib have been compared to those on placebo and non-specific inhibitors of both COX-1 and COX-2. In 12-week endoscopy studies celecoxib (100 mg - 800 mg/day) was associated with a significantly lower risk of gastroduodenal ulcers compared with naproxen (1,000 mg/day) and ibuprofen (2400 mg/day). The data were inconsistent in comparison with diclofenac (150 mg/day). In 2 of the 12-week studies, the percentage of patients with endoscopic gastroduodenal ulceration was not significantly different between placebo and celecoxib 200 mg twice daily and 400 mg twice daily.
Table 2 summarizes the incidence of endoscopic ulcers in two 12-week studies that enrolled patients in whom baseline endoscopies revealed no ulcers. (See Table 2.)
Click on icon to see table/diagram/image
Table 3 summarizes data from two 12-week studies that enrolled patients in whom baseline endoscopies revealed no ulcers. Patients underwent interval endoscopies every 4 weeks to give information on ulcer risk over time. (See Table 3.)
Click on icon to see table/diagram/image
One randomized and double-blind 6-month study in 430 RA patients was conducted in which an endoscopic examination was performed at 6 months.
The incidence of endoscopic ulcers in patients taking celecoxib 200 mg twice daily was 4% vs. 15% for patients taking diclofenac SR 75 mg twice daily (p<0.001).
In 4 of the 5 endoscopic studies, approximately 11% of patients (440/4,000) were taking aspirin (≤325 mg/day). In the celecoxib groups, the endoscopic ulcer rate appeared to be higher in aspirin users than in non-users. However, the increased rate of ulcers in these aspirin users was less than the endoscopic ulcer rates observed in the active comparator groups, with or without aspirin.
The correlation between findings of endoscopic studies, and the relative incidence of clinically significant serious upper GI events has not been established. Serious clinically significant upper GI bleeding has been observed in patients receiving celecoxib in controlled and open-labeled trials, albeit infrequently (see Precautions).
Gastrointestinal Safety Meta-Analysis from Osteoarthritis and Rheumatoid Arthritis Studies: An analysis of 31 randomized controlled clinical studies in OA and RA, involving 39,605 patients with OA (N = 5,903), RA (N = 3,232), or patients with either condition (N = 10,470) compared the incidence of GI adverse events in celecoxib treated patients to the incidence in patients administered placebo or NSAIDs (including naproxen, diclofenac and ibuprofen). The incidence of clinical ulcers and ulcer bleeds with celecoxib 200 mg to 400 mg total daily dose was 0.2% compared to an incidence of 0.6% with NSAIDs (RR = 0.35; 95% CI 0.22 - 0.56).
Cardiovascular Safety - Long-term Studies Involving Patients with Sporadic Adenomatous Polyps: Two studies involving patients with sporadic adenomatous polyps were conducted with celecoxib, i.e., the APC trial (Adenoma Prevention with Celecoxib) and the PreSAP trial (Prevention of Spontaneous Adenomatous Polyps). In the APC trial, there was a dose-related increase in the composite endpoint of CV death, myocardial infarction, or stroke (adjudicated) with celecoxib compared to placebo over 3 years of treatment. The PreSAP trial did not demonstrate a statistically significant increased risk for the same composite endpoint.
In the APC trial, the hazard ratios compared to placebo for a composite endpoint of CV death, myocardial infarction, or stroke (adjudicated) were 3.4 (95% CI 1.4 - 8.5) with celecoxib 400 mg twice daily, and 2.8 (95% CI 1.1 - 7.2) with celecoxib 200 mg twice daily, cumulative rates for this composite endpoint over 3 years were 3.0% (20/671) and 2.5% (17/685) for 400 mg twice daily and 200 mg twice daily celecoxib treatment groups, respectively, compared to 0.9% (6/679) for placebo group. The increases for both celecoxib dose groups versus placebo were mainly driven by myocardial infarction.
In the PreSAP trial, the hazard ratio compared to placebo for this same composite endpoint was 1.2 (95% CI 0.6 - 2.4) with celecoxib 400 mg once daily. Cumulative rate for this composite endpoint over 3 years was 2.3% (21/933), compared to 1.9% (12/628) for placebo group.
Cardiovascular Safety - Long-term Study of Alzheimer's Disease Anti-inflammatory Prevention Trial (ADAPT): Data from the ADAPT study did not show a significantly increased CV risk with celecoxib 200 mg twice daily compared to placebo. The relative risk compared to placebo for a similar composite endpoint (CV death, MI, stroke) was 1.14 (95% CI 0.61 - 2.15) with celecoxib 200 mg twice daily.
Cardiovascular Safety - Meta-Analysis from Chronic Usage Studies: A meta-analysis of safety data (adjudicated, investigator-reported serious adverse events) from 39 completed celecoxib clinical studies of up to 65 weeks duration has been conducted, representing 41,077 patients: [23,030 (56.1%) patients exposed to celecoxib 200 mg to 800 mg total daily dose (TDD); 13,990 (34.1%) patients exposed to non-selective NSAIDs; and 4,057(9.9%) patients exposed to placebo].
In this analysis, the adjudicated event rate for the composite endpoint of CV death, non-fatal myocardial infarction and non-fatal stroke was similar between celecoxib (N = 19,773; 0.96 events/100 patient-years) and non-selective NSAIDs (N = 13,990; 1.12 events/100 patient-years) treatment (RR = 0.90, 95% CI 0.60 - 1. 33). This pattern of effect was maintained with or without ASA use (≤325 mg). The adjudicated event rate of non-fatal myocardial infarction trended higher (RR = 1.76, 95% CI 0.93 - 3.35); however, that of non-fatal stroke trended lower (RR = 0.51, 95% CI 0.23 - 1.10), and that of CV death was comparable (RR = 0.57, 95% CI 0.28 - 1.14) for celecoxib compared to combined non-selective NSAIDs.
In this analysis, the adjudicated event rate for the composite endpoint of CV death, non-fatal myocardial infarction and non-fatal stroke was 1.42/100 patient-years for celecoxib (N = 7,462) and 1.20/100 patient-years for placebo (N = 4,057) treatment (RR = 1.11, 95% CI 0.47 - 2.67). This pattern of effect was maintained with or without ASA use (≤325 mg). The incidence of non-fatal myocardial infarction trended higher (RR = 1.56, 95% CI 0.21 - 11.90), as did that of CV death (RR = 1.26, 95% CI 0.33 - 4.77), and that of non-fatal stroke was similar (RR = 0.80, 95% CI 0.19 - 3.31) for celecoxib compared to placebo.
Cardiovascular Safety: CV safety outcomes were evaluated in the CLASS trial (see description of trial as previously mentioned). Kaplan-Meier cumulative rates for investigator-reported serious CV thromboembolic adverse events (including MI, pulmonary embolism, deep venous thrombosis, unstable angina, transient ischemic attacks, and ischemic cerebrovascular accidents) demonstrated no differences between the celecoxib, diclofenac, or ibuprofen treatment groups. The cumulative rates in all patients at nine months for celecoxib, diclofenac, and ibuprofen were 1.2%, 1.4%, and 1.1%, respectively. The cumulative rates in non-ASA users at nine months in each of the three treatment groups were less than 1%. The cumulative rates for myocardial infarction in non-ASA users at nine months in each of the three treatment groups were less than 0.2%. There was no placebo group in the CLASS trial, which limits the ability to determine whether the three drugs tested had no increased risk of CV events or if they all increased the risk to a similar degree.
The Celecoxib Long-term Arthritis Safety Study (CLASS) Including Use with Aspirin: In a prospective long-term safety outcome study conducted post-marketing in approximately 5,800 OA patients and 2,200 RA patients, patients received celecoxib 400 mg twice daily (4-fold and 2-fold the recommended OA and RA doses, respectively), ibuprofen 800 mg three times daily, or diclofenac 75 mg twice daily (common therapeutic doses). Median exposures for celecoxib (n = 3,987) and diclofenac (n = 1,996) were 9 months while ibuprofen (n = 1,985) was 6 months. The Kaplan-Meier cumulative rates at 9 months are provided for all analyses. The primary endpoint of this outcome study was the incidence of complicated ulcers (gastrointestinal bleeding, perforation, or obstruction).
Patients were allowed to take concomitant low-dose (≤325 mg/day) aspirin (ASA) for CV prophylaxis (ASA subgroups: celecoxib, n = 882; diclofenac, n = 445; ibuprofen, n = 412). Differences in the incidence of complicated ulcers between celecoxib and the combined group of ibuprofen and diclofenac were not statistically significant. Those patients on celecoxib and concomitant low-dose ASA experienced 4-fold higher rates of complicated ulcers compared to those not on ASA (see Precautions). The results for celecoxib are displayed in Table 4. (See Table 4.)
Click on icon to see table/diagram/image
Platelet Function: In healthy volunteers, celecoxib at therapeutic doses and at multiple doses of 600 mg twice daily (three times the highest recommended dose) had no effect on platelet aggregation and bleeding time compared to placebo. Active controls (non-specific COX inhibitors) all significantly reduced platelet aggregation and prolonged bleeding time (see figure).
Click on icon to see table/diagram/image
Celecoxib versus Omeprazole and Diclofenac for At-Risk Osteoarthritis and Rheumatoid Arthritis Patients (CONDOR) trial: In a prospective randomised 24-week safety study in patients who were aged ≥60 years or had a history of gastroduodenal ulcers (users of ASA excluded), the percentages of patients with decreases in hemoglobin (≥2 g/dL) and/or hematocrit (≥10%) of defined or presumed GI origin were lower in patients treated with celecoxib 200 mg twice daily (N = 2,238) compared to patients treated with diclofenac SR 75 mg twice daily plus omeprazole 20 mg once daily (N = 2,246) [0.2% vs. 1.1% for defined GI origin, p = 0.004; 0.4% vs. 2.4% for presumed GI origin, p = 0.0001]. The rates of clinically detected GI complications such as perforation, obstruction, or hemorrhage were very low with no differences between the treatment groups (4-5 per group). Results for the individual components of this composite endpoint were as follows. (See Table 5.)
Click on icon to see table/diagram/image
Prospective Randomized Evaluation of Celecoxib Integrated Safety vs. Ibuprofen Or Naproxen (PRECISION): Design: The PRECISION study was a double-blind study of cardiovascular safety in OA or RA patients with or at high risk for cardiovascular disease comparing Celecoxib (200-400 mg daily) with Naproxen (750-1000 mg daily) and Ibuprofen (1800-2400 mg daily). The primary endpoint, Antiplatelet Trialists Collaboration (APTC), was an independently adjudicated composite of cardiovascular death (including hemorrhagic death), non-fatal myocardial infarction or non-fatal stroke. The study was
planned with 80% power to evaluate non-inferiority. All patients were prescribed open-label esomeprazole (20-40 mg) for gastro protection. Patients who were taking low-dose Aspirin were permitted to continue therapy.
Other independently adjudicated secondary and tertiary endpoints included cardiovascular, gastrointestinal and renal outcomes. Additionally, there was a 4-month substudy focusing on the effects of the three drugs on blood pressure as measured by ambulatory monitoring (ABPM).
Results: (See Table 6.)
Click on icon to see table/diagram/image
Primary Endpoint: Celecoxib, as compared with either naproxen or ibuprofen, met all four prespecified non-inferiority requirements (p<0.001 for non-inferiority in both comparisons). Non-inferiority is established when the hazard ratio (HR) ≤1.12 in both ITT and mITT analyses, and upper 95% CI ≤1.33 for ITT analysis and ≤1.40 for mITT analysis.
The primary analysis for ITT and mITT are described as follows in Table 7. (See Table 7.)
Click on icon to see table/diagram/image
Key Secondary and Tertiary Endpoints: The analysis of Major Adverse Cardiovascular Events (MACE)* for mITT are described as follows in Table 8. (See Table 8.)
Click on icon to see table/diagram/image
The analysis of Gastrointestinal Events for mITT are described as follows in Table 9. (See Table 9.)
Click on icon to see table/diagram/image
In the ITT population for the CSGIE endpoint there were no significant differences, in the pairwise comparisons between treatment regimens (data not shown). For the endpoint of iron deficiency anemia of GI origin, significant differences (celecoxib vs. naproxen; celecoxib vs. ibuprofen) and nonsignificant differences (ibuprofen vs. naproxen) were observed in a manner consistent with the data presented previously.
The analysis of clinically significant renal events*, hospitalization for CHF and hypertension for mITT are described as follows in Table 10. (See Table 10.)
Click on icon to see table/diagram/image
In the ITT population for the endpoint of clinically significant renal events, only the pairwise comparison between celecoxib and ibuprofen was significant, HR 0.61 (0.44, 0.85), no significant differences were observed between treatment regimens in the incidence of hospitalization for congestive heart failure, and a significantly lower incidence of hospitalization for hypertension was observed between celecoxib and ibuprofen, HR 0.59 (0.36, 0.99).
All-cause mortality: In the mITT populations celecoxib, naproxen and ibuprofen were associated with 53 (0.7%), 79 (1.0%), and 73 (0.9%) deaths, respectively. Significant differences were observed in the pairwise comparisons between celecoxib and either naproxen HR 0.65 (0.46, 0.92) or celecoxib and ibuprofen HR 0.68 (0.48, 0.97). In the ITT population the celecoxib, naproxen and ibuprofen were associated with 132 (1.6%), 163 (2.0%) and 142 (1.8%) deaths, respectively. No significant differences were observed in pairwise comparisons between treatments.
ABPM Substudy: In the PRECISION-ABPM substudy, among the total of 444 analyzable patients, at Month 4, celecoxib-treated patients had the smallest change in 24-hour ambulatory systolic blood pressure
(SBP) compared to ibuprofen and naproxen: celecoxib produced a slight reduction of 0.3 mmHg while ibuprofen and naproxen increased mean 24-hour SBP by 3.7 and 1.6 mmHg, respectively. These changes resulted in a statistically significant and clinically meaningful difference of -3.9 mmHg (p=0.0009) between celecoxib and ibuprofen; a non-significant difference of -1.8 (p=0.119) mmHg between celecoxib and naproxen, and a non-significant difference of -2.1 mmHg (p=0.0787) between naproxen and ibuprofen.
Pharmacokinetics: Absorption: The pharmacokinetics of celecoxib has been evaluated in approximately 1500 individuals. When given under fasting conditions celecoxib is well absorbed reaching peak plasma concentrations after approximately 2-3 hours. Oral bioavailability from capsules is about 99% relative to administration in suspension (optimally available oral dosage form). Under fasting conditions, both peak plasma levels (Cmax) and area under the curve (AUC) are roughly dose proportional up to 200 mg twice daily; at higher doses there are less than proportional increases in Cmax and AUC.
Distribution: Plasma protein binding, which is concentration independent, is about 97% at therapeutic plasma concentrations and celecoxib is not preferentially bound to erythrocytes in the blood.
Metabolism: Celecoxib metabolism is primarily mediated via cytochrome P450 2C9. Three metabolites, inactive as COX-1 or COX-2 inhibitors, have been identified in human plasma; a primary alcohol, the corresponding carboxylic acid and its glucuronide conjugate.
Cytochrome P450 2C9 activity is reduced in individuals with genetic polymorphisms that lead to reduced enzyme activity, such as those homozygous for the CYP2C9*3 polymorphism.
In a pharmacokinetic study of celecoxib 200 mg administered once daily in healthy volunteers, genotyped as either CYP2C9*1/*1, CYP2C9*1/*3, or CYP2C9*3/*3, the median Cmax and AUC0-24 of celecoxib on day 7 were approximately 4-fold and 7-fold, respectively, in subjects genotyped as CYP2C9*3/*3 compared to other genotypes. In three separate single-dose studies, involving a total of 5 subjects genotyped as CYP2C9*3/*3, single-dose AUC0-24 increased by approximately 3-fold compared to normal metabolizers. It is estimated that the frequency of the homozygous *3/*3 genotype is 0.3% - 1.0% among different ethnic groups.
Patients who are known, or suspected to be CYP2C9 poor metabolizers based on previous history/experience with other CYP2C9 substrates should be administered celecoxib with caution. Consider starting treatment at half the lowest recommended dose (see Dosage & Administration).
Excretion: Elimination of celecoxib is mostly by hepatic metabolism with less than 1% of the dose excreted unchanged in urine. After multiple dosing, elimination half-life is 8 to 12 hours and the rate of clearance is about 500 mL/min. With multiple dosing, steady-state plasma concentrations are reached before day 5. The intersubject variability on the main pharmacokinetic parameters (AUC, Cmax,
elimination half-life) is about 30%. The mean steady-state volume of distribution is about 500 L/70 kg in young healthy adults indicating wide distribution of celecoxib into the tissues. Pre-clinical studies indicate that the drug crosses the blood/brain barrier.
Food Effects: Dosing with food (high fat meal) delays absorption of celecoxib resulting in a Tmax of about 4 hours and increases bioavailability by about 20% (see Dosage & Administration).
In healthy adult volunteers, the overall systemic exposure (AUC) of celecoxib was equivalent when celecoxib was administered as intact capsule or capsule contents sprinkled on applesauce. There were no significant alterations in Cmax, Tmax or T½ after administration of capsule contents on applesauce.
Special Populations: Elderly: In the population >65 years, there is a one and a half to two-fold increase in mean Cmax and AUC for celecoxib. This is a predominantly weight-related rather than age-related change, celecoxib levels being higher in lower weight individuals and consequently higher in the elderly population who are generally of lower mean weight than the younger population. Therefore, elderly females tend to have higher drug plasma concentrations than elderly males. No dosage adjustment is generally necessary. However, for elderly patients with a lower than average body weight (<50 kg), therapy should be initiated at the lowest recommended dose.
Race: A meta-analysis of pharmacokinetic studies has suggested an approximately 40% higher AUC of celecoxib in the Black population compared to Caucasians.
The cause and clinical significance of this finding is unknown and therefore treatment should be initiated at the lowest recommended dose.
Hepatic Impairment: Plasma concentrations of celecoxib in patients with mild hepatic impairment (Child-Pugh Class A) are not significantly different from those of age and sex matched controls. In patients with moderate hepatic impairment (Child-Pugh Class B) celecoxib plasma concentrations are about twice those of matched controls. No dosage adjustment is necessary in patients with mild hepatic impairment. Treatment should be initiated at half the recommended dose in patients with moderate liver impairment (with serum albumin 25-35 g/L or Child-Pugh Class B) (see Dosage & Admnistration).
There is no clinical experience in patients with severe hepatic impairment. The use of celecoxib in patients with severe hepatic impairment is not recommended (see Precautions).
Renal Impairment: In elderly volunteers with age related reductions in glomerular filtration rate (GFR) (mean GFR>65 mL/min/1.73 m
2) and in patients with chronic stable renal insufficiency (GFR 35-60 mL/min/1.73 m
2) celecoxib pharmacokinetics was comparable to those seen in patients with normal renal function. No significant relationship was found between serum creatinine (or creatinine clearance) and celecoxib clearance. Severe renal insufficiency would not be expected to alter clearance of celecoxib since the main route of elimination is via hepatic metabolism to inactive metabolites.
Renal Effects: The relative roles of COX-1 and COX-2 in renal physiology are not completely understood. Celecoxib reduces the urinary excretion of PGE2 and 6-keto-PGF1α (a prostacyclin metabolite) but leaves serum thromboxane B2 (TXB2) and urinary excretion of 11-dehydro-TXB2, a thromboxane metabolite (both COX-1 products) unaffected. Specific studies have shown celecoxib produces no decreases in GFR in the elderly or those with chronic renal insufficiency. These studies have also shown transient reductions in fractional excretion of sodium. In studies in patients with arthritis a comparable incidence of peripheral edema has been observed to that seen with non-specific COX inhibitors (which also possess COX-2 inhibitory activity). This was most evident in patients receiving concomitant diuretic therapy. However, increased incidences of hypertension and cardiac failure have not been observed and the peripheral edema has been mild and self-limiting.
Toxicology: Preclinical safety data: Preclinical safety data revealed no special hazard for humans based on conventional studies of repeated dose toxicity, mutagenicity or carcinogenicity.
Celecoxib at oral doses ≥150 mg/kg/day (approximately 2-fold human exposure at 200 mg twice daily as measured by AUC0-24), caused an increased incidence of ventricular septal defects, a rare event,
and fetal alterations, such as ribs fused, sternebrae fused and sternebrae misshapen when rabbits were treated throughout organogenesis. A dose-dependent increase in diaphragmatic hernias was observed
when rats were given celecoxib at oral doses ≥30 mg/kg/day (approximately 6-fold human exposure based on the AUC0-24 at 200 mg twice daily) throughout organogenesis. These effects are expected
following inhibition of prostaglandin synthesis. In rats, exposure to celecoxib during early embryonic development resulted in pre-implantation and post-implantation losses, and reduced embryo/fetal survival.
Animal Toxicology: An increase in the incidence of background findings of spermatocele with or without secondary changes, such as epididymal hypospermia as well as minimal to slight dilation of the seminiferous tubules was seen in the juvenile rat. These reproductive findings while apparently treatment-related did not increase in incidence or severity with dose and may indicate an exacerbation of a spontaneous condition. Similar reproductive findings were not observed in studies of juvenile or adult dogs or in adult rats treated with celecoxib. The clinical significance of this observation is unknown.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image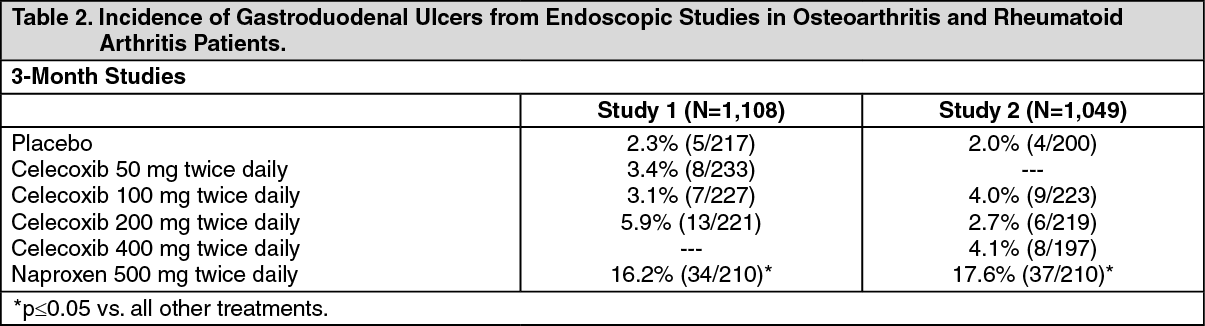 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image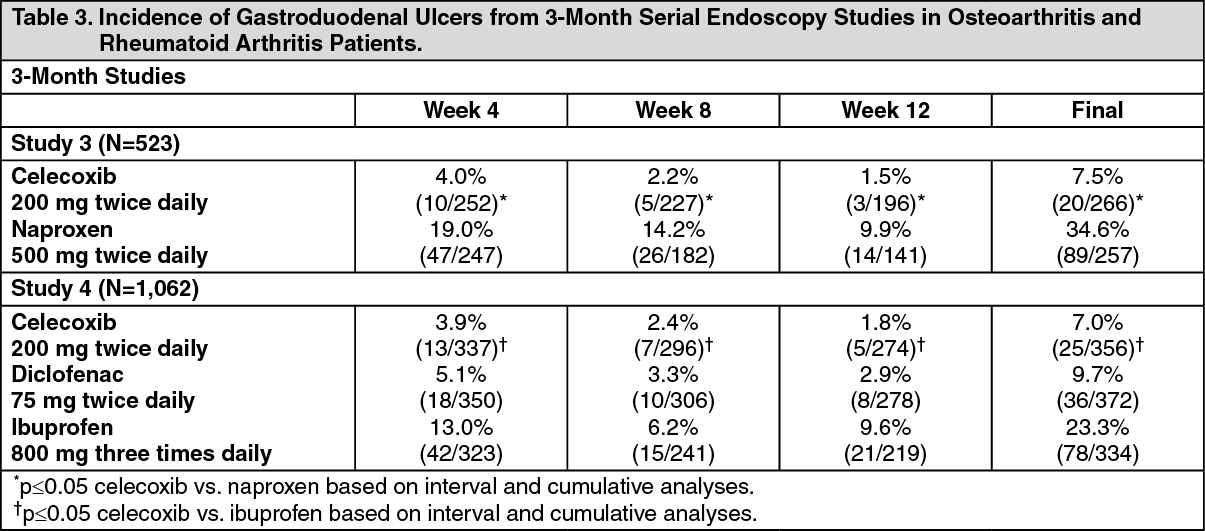 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image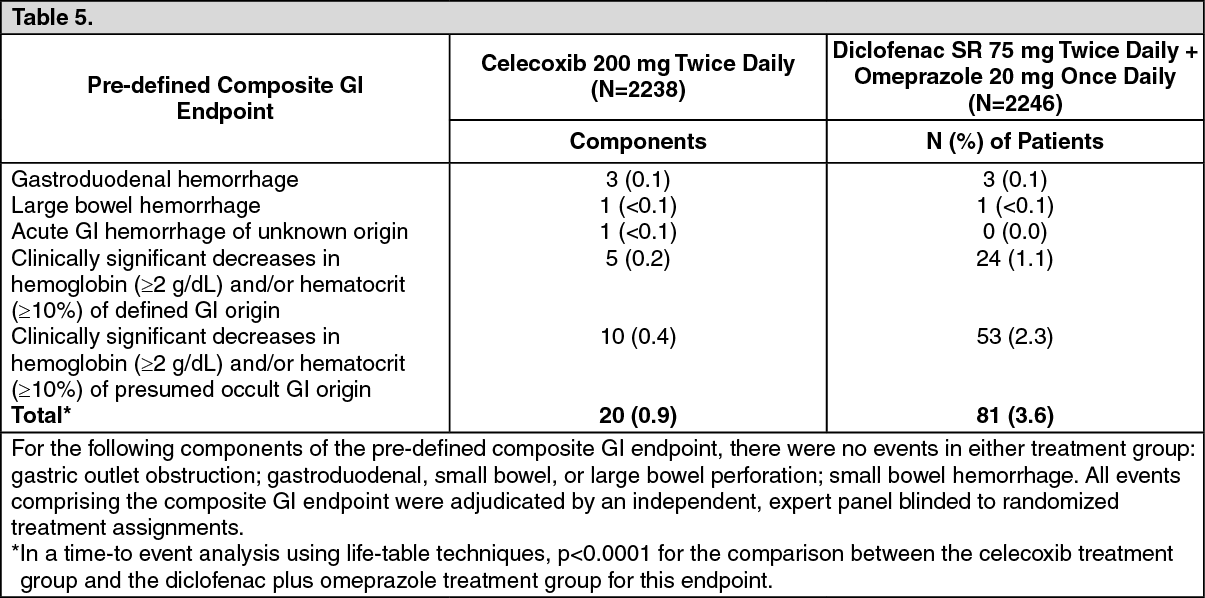 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image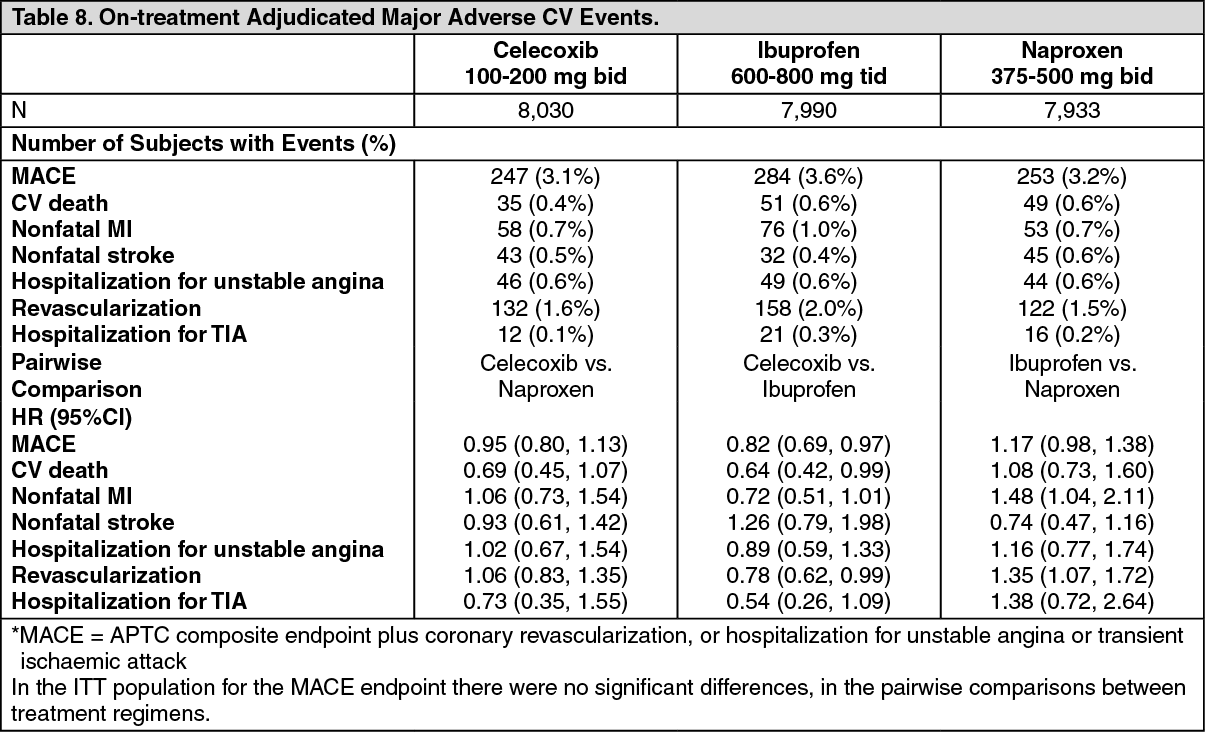 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image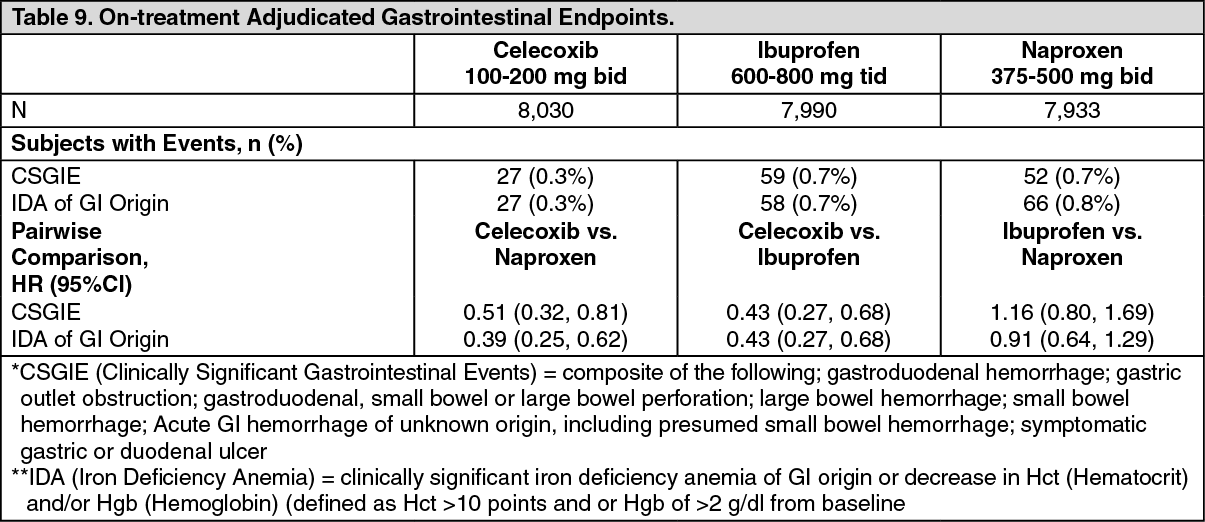 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image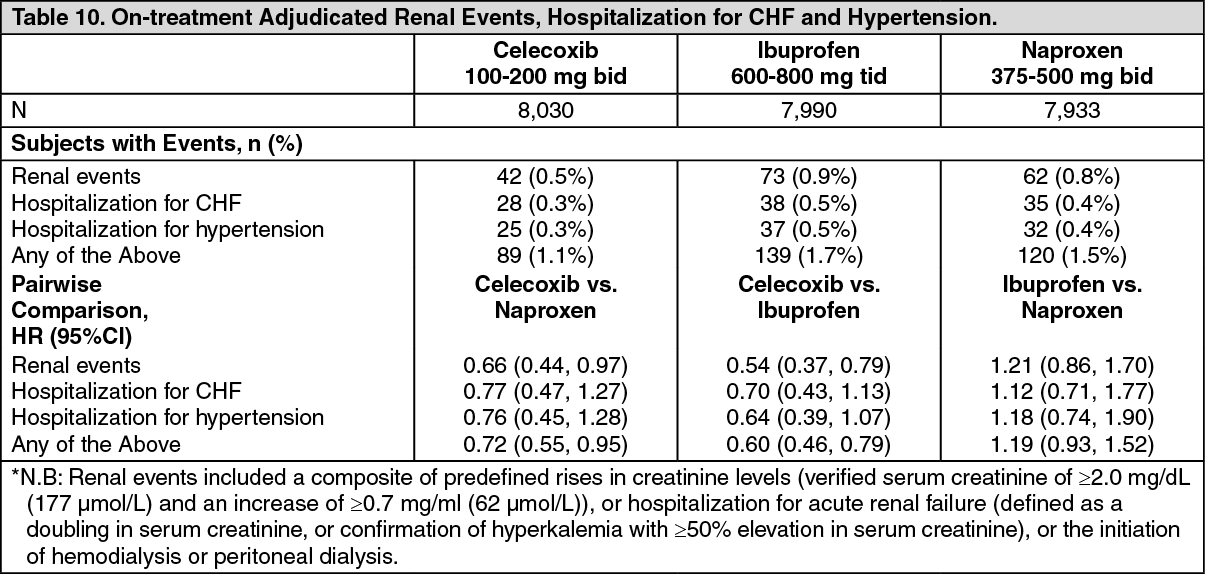 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image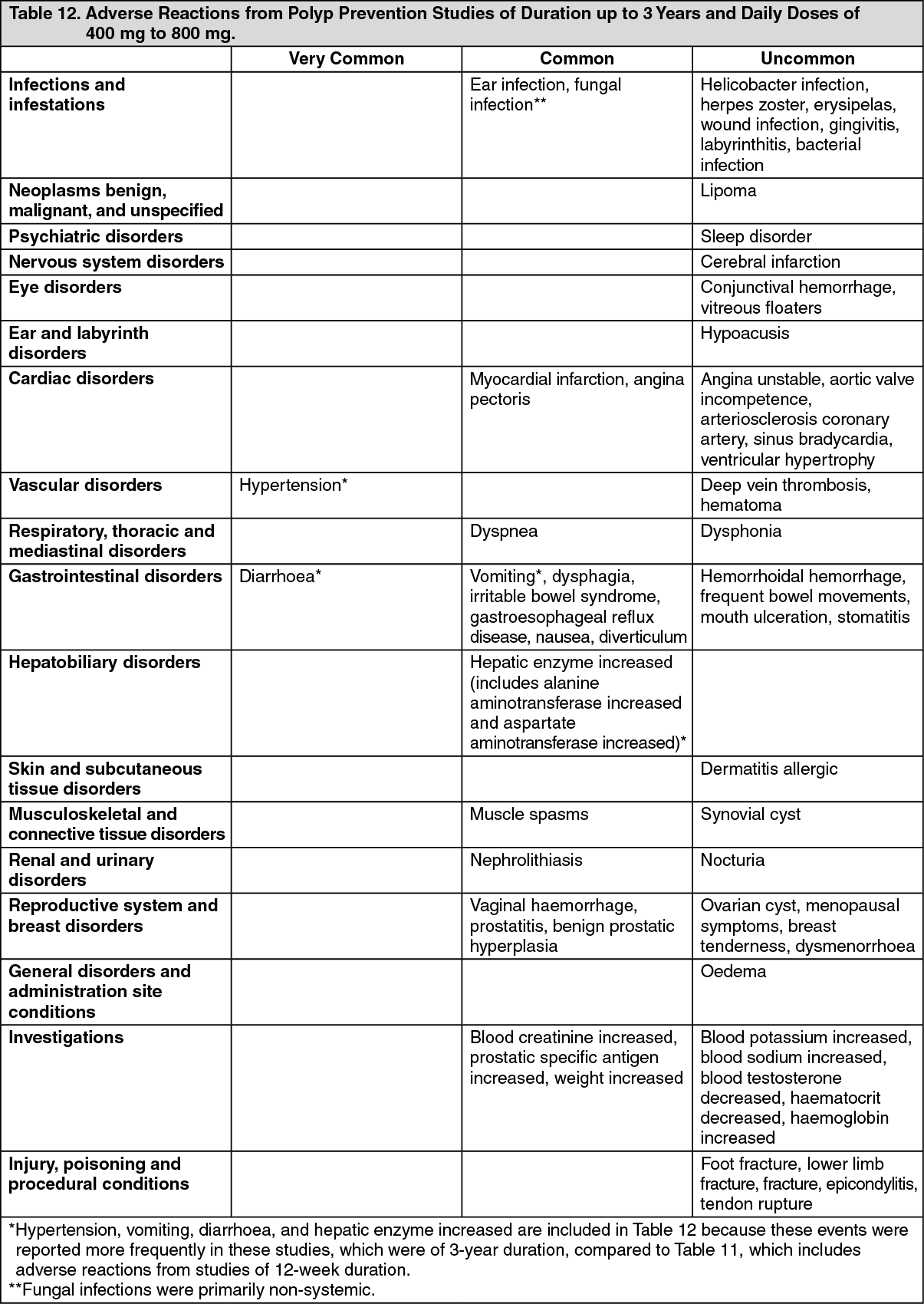 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image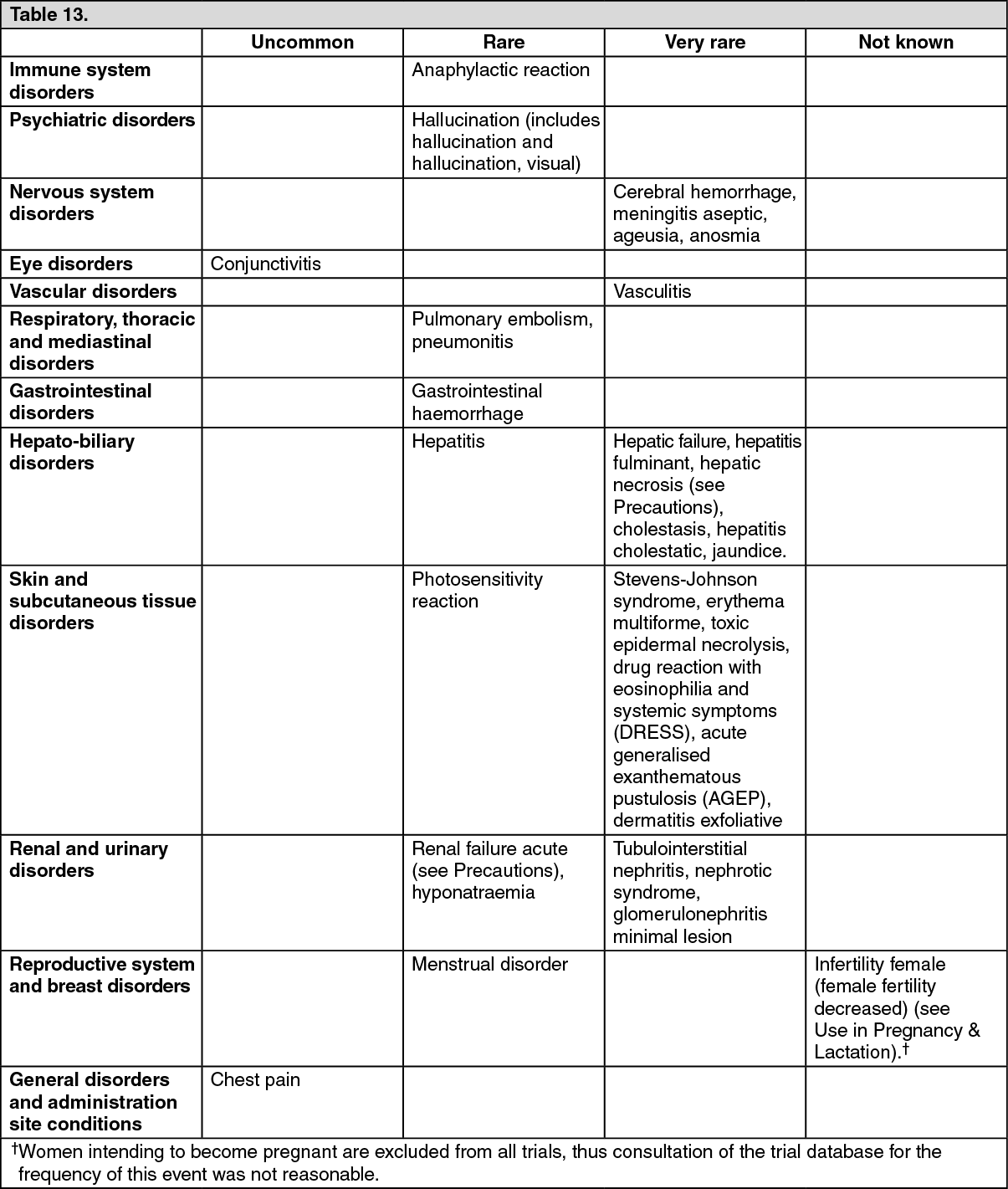 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
