Pharmacotherapeutic group: Aldosterone Antagonists.
ATC Code: C03DA05.
Pharmacodynamics: Mechanism of action: Finerenone is a nonsteroidal, selective antagonist of the mineralocorticoid receptor (MR) that potently attenuates inflammation and fibrosis mediated by MR overactivation. The MR is expressed in the kidneys, heart and blood vessels where finerenone also counteracts sodium retention and hypertrophic processes. Finerenone has a high potency and selectivity for the MR due to its nonsteroidal structure and bulky binding mode. Finerenone has no relevant affinity for androgen, progesterone, estrogen and glucocorticoid receptors and therefore does not cause sex hormone-related adverse events (e.g., gynecomastia). Its binding to the MR leads to a specific receptor ligand complex that blocks recruitment of transcriptional coactivators implicated in the expression of pro-inflammatory and pro-fibrotic mediators.
Pharmacodynamic effects: Effects in healthy participants: Multiple dose regimens of finerenone (daily doses of 20 mg or 40 mg over 10 days) led to activation of the renin-angiotensin-aldosterone system (RAAS), i.e., reversible increases of plasma renin activity and serum aldosterone concentrations with baseline values reached again within 48 hours after the last dose.
Following activation of the MR with the agonist fludrocortisone single doses of finerenone up to 20 mg showed dose dependent natriuretic effects while decreasing urinary potassium excretion as compared to placebo.
Single or multiple doses of finerenone did not influence vital signs parameters in healthy participants.
Effects in patients with CKD and T2D: In FIDELIO-DKD and FIGARO-DKD, randomized, double-blind, placebo-controlled, multicenter phase III studies in adults with CKD and T2D, the placebo-corrected relative reduction in urinary albumin-to-creatinine ratio (UACR) in patients randomized to finerenone at Month 4 was 31% and 32%, respectively and UACR remained reduced throughout both studies.
In ARTS DN, a randomized, double-blind, placebo-controlled, multicenter phase IIb dose-finding study in adults with CKD and T2D, the placebo-corrected relative reduction in UACR at Day 90 was 25% and 38% in patients treated with finerenone 10 mg and 20 mg once daily, respectively.
Cardiac electrophysiology: In a thorough QT study in 57 healthy participants, there was no indication of a QT/QTc prolonging effect of finerenone after single doses of 20 mg (therapeutic) or 80 mg (supratherapeutic), indicating that finerenone has no effect on cardiac repolarization.
Clinical efficacy and safety: Chronic kidney disease and type 2 diabetes: Finerenone (Firialta) was investigated in two randomized, double-blind, placebo-controlled, multicenter phase III studies, FIDELIO-DKD and FIGARO-DKD. In these studies, the effect of Finerenone (Firialta) on kidney and cardiovascular outcomes was evaluated in adults with CKD and T2D receiving either Finerenone (Firialta) 10 mg or 20 mg once daily, or placebo.
In FIDELIO-DKD patients were eligible based on evidence of persistent albuminuria (>30 mg/g to 5,000 mg/g), an eGFR of 25 to 75 mL/min/1.73 m
2, serum potassium ≤4.8 mmol/L at screening, and were required to be receiving standard of care, including a maximum tolerated labeled dose of an angiotensin-converting enzyme inhibitor (ACEi) or angiotensin receptor blocker (ARB).
The primary endpoint in the FIDELIO-DKD study was a composite of time to first occurrence of kidney failure (defined as chronic dialysis or kidney transplantation, or a sustained decrease in eGFR to <15 mL/min/1.73 m
2 over at least 4 weeks), a sustained decline in eGFR of 40% or more compared to baseline over at least 4 weeks, or renal death. The key secondary endpoint was a composite of time to first occurrence of cardiovascular (CV) death, non-fatal myocardial infarction (MI), non-fatal stroke or hospitalization for heart failure.
The trial analyzed 5,674 patients randomly assigned to receive either Finerenone (Firialta) (N=2833), or placebo (N=2841), with a median follow-up duration of 2.6 years. After the end of study notification, vital status was obtained for 99.7% of patients. The trial population was 63% White, 25% Asian and 5% Black. The mean age at enrollment was 66 years and 70% of patients were male. At baseline, the mean eGFR was 44.3 ml/min/1.73 m
2, with 55% of patients having an eGFR <45 mL/min/1.73 m
2, median urine albumin-to-creatinine ratio (UACR) was 852 mg/g, and mean glycated hemoglobin A1c (HbA1c) was 7.7%, 46% had a history of atherosclerotic cardiovascular disease, 30% had history of coronary artery disease, 8% had a history of cardiac failure, and the mean blood pressure was 138/76 mmHg. The mean duration of type 2 diabetes at baseline was 16.6 years and a history of diabetic retinopathy and diabetic neuropathy was reported in 47% and 26% of patients, respectively. At baseline, almost all patients were on ACEi (34%) or ARB (66%), and 97% of patients used one or more antidiabetic medications (insulin [64%], biguanides [44%], glucagon-like peptide-1 [GLP-1] receptor agonists [7%], sodium-glucose cotransporter 2 [SGLT2] inhibitors [5%]). The other most frequent medications taken at baseline were statins (74%) and calcium channel blockers (63%).
Finerenone (Firialta) demonstrated superiority to placebo by significantly reducing the risk of the primary composite endpoint compared to placebo in a time-to-event analysis using the Cox proportional hazards model and log-rank test (HR 0.82, 95% CI 0.73-0.93, p=0.0014). (See Figure 1/Table 1 as follows.) Finerenone (Firialta) also significantly reduced the risk of the key secondary composite endpoint of time to first occurrence of CV death, non-fatal MI, non-fatal stroke or hospitalization for heart failure compared to placebo (HR 0.86, 95% CI 0.75-0.99, p=0.0339). (See Figure 2.) Prespecified secondary time-to-event endpoints are included in Table 1. The treatment effect for the primary and key secondary endpoints was generally consistent across subgroups, including region, eGFR, UACR, systolic blood pressure (SBP) and HbA1c at baseline.
In the FIDELIO-DKD study, hyperkalemia events were reported in 18.3% of Finerenone (Firialta)-treated patients compared with 9.0% of placebo-treated patients. Hospitalization due to hyperkalemia for the Finerenone (Firialta) group was 1.4% versus 0.3% in the placebo group. Hyperkalemia leading to permanent discontinuation in patients who received Finerenone (Firialta) was 2.3% versus 0.9% in the placebo group.
In the FIDELIO-DKD study, Glomerular filtration rate decreased events were reported in 6.3% of Finerenone (Firialta)-treated patients compared with 4.7% of placebo-treated patients, and those leading to permanent discontinuation in patients receiving Finerenone (Firialta) were 0.2% versus 0.3% in the placebo group. Patients on Finerenone (Firialta) experienced an initial decrease in eGFR (mean 2 mL/min/1.73 m
2) that attenuated over time compared to placebo. This decrease was reversible after treatment discontinuation. The initial decrease in eGFR was associated with long term preservation of kidney function.
The FIGARO-DKD study included adults with CKD and T2D, based on having a UACR of ≥30 mg/g to <300 mg/g and an eGFR of 25 to 90 mL/min/1.73 m
2, or a UACR ≥300 mg/g and an eGFR ≥60 mL/min/1.73 m
2 at screening. Patients were required to have a serum potassium of ≤4.8 mmol/L at screening and received standard of care, including a maximum tolerated labeled dose of a RAS inhibitor (either an ACEi or ARB).
The primary endpoint in the FIGARO-DKD study was a composite of time to first occurrence of CV death, non-fatal MI, non-fatal stroke or hospitalization for heart failure. Secondary endpoints included a composite of time to kidney failure, a sustained decline in eGFR of 40% or more compared to baseline over at least 4 weeks, or renal death and a composite of time to kidney failure, a sustained decline in eGFR of 57% or more compared to baseline, or renal death.
The trial analyzed 7,352 patients randomly assigned to receive either Finerenone (Firialta) (N=3686), or placebo (N=3666) that were followed for a median duration of 3.4 years. After the end of study notification, vital status was obtained for 99.8% of patients. The trial population was 72% White, 20% Asian and 4% Black. The mean age at enrollment was 64 years and 69% of patients were male. At baseline, the mean eGFR was 67.8 mL/min/1.73 m
2, with 62% of patients having an eGFR ≥60 mL/min/1.73 m
2, median UACR was 308 mg/g, and mean glycated HbA1c was 7.7%, 45% of patients had a history of atherosclerotic cardiovascular disease, 8% had a history of cardiac failure, and the mean blood pressure was 136/77 mmHg. The mean duration of type 2 diabetes at baseline was 14.5 years and a history of diabetic retinopathy and diabetic neuropathy was reported in 31% and 28% of patients, respectively. At baseline, almost all patients were on a RAS-inhibitor and 98% of patients used one or more antidiabetic medications (insulin [54%], biguanides [69%], GLP-1 receptor agonists [7%], SGLT2 inhibitors [8%]). The other most frequent medication class taken at baseline was statins (71 %).
Finerenone (Firialta) significantly reduced the risk of the primary composite endpoint compared to placebo in a time to event analysis using the Cox proportional hazards model and log rank test (HR 0.87, 95% CI 0.76-0.98, p=0.0264). (See Figure 3/Table 2 as follows.) The treatment effect for the primary endpoint was consistent across subgroups, including region, eGFR, UACR, SBP and HbA1c at baseline. A lower incidence rate of the secondary composite outcome of kidney failure, sustained eGFR decline of 40% or more or renal death was observed in the Finerenone (Firialta) group compared to placebo, however this difference did not achieve statistical significance (HR 0.87, 95% CI 0.76-1.01, p=0.0689). (See Figure 4/Table 2 as follows.) A lower risk of the secondary outcome of kidney failure, sustained eGFR decline of 57% or more or renal death was observed in the Finerenone (Firialta) group compared to placebo (HR 0.77, 95% CI 0.60-0.99). Prespecified secondary time-to-event endpoints are included in Table 2.
In the FIGARO-DKD study, hyperkalemia events were reported in 10.8% of Finerenone (Firialta)-treated patients compared with 5.3% of placebo-treated patients. Hospitalization due to hyperkalemia for the Finerenone (Firialta) group was 0.6% versus <0.1% in the placebo group. Hyperkalemia leading to permanent discontinuation in patients who received Finerenone (Firialta) was 1.2% versus 0.4% in the placebo group.
In the FIGARO-DKD study, Glomerular filtration rate decreased events were reported in 4.6% of Finerenone (Firialta)-treated patients compared with 3.9% of placebo-treated patients, and those leading to permanent discontinuation in patients receiving Finerenone (Firialta) were 0.2% versus 0.1% in the placebo group. Patients on Finerenone (Firialta) experienced an initial decrease in eGFR of around 2 mL/min/1.73 m
2 that attenuated over time compared to placebo. This decrease was reversible after treatment discontinuation. The initial decrease in eGFR was associated with long term preservation of kidney function. (See Tables 1 and 2 and Figures 1, 2, 3 and 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In a pre-specified pooled analysis of the FIDELIO-DKD and FIGARO-DKD studies, finerenone reduced the risk of the CV composite endpoint of time to CV death, non-fatal MI, non-fatal stroke or hospitalization for heart failure compared to placebo (HR 0.86 [95% CI 0.78; 0.95]). (See Figure 5.) The risk of the kidney composite endpoint of time to kidney failure, a sustained decrease in eGFR of 40% or more compared to baseline or renal death was also reduced with finerenone compared to placebo (HR 0.85 [95% CI 0.77; 0.93]), as was the composite endpoint of time to kidney failure, a sustained decrease in eGFR of 57% or more compared to baseline or renal death (HR 0.77 [95% CI 0.67; 0.88]). (See Figure 5.)
Click on icon to see table/diagram/image
Pharmacokinetics: Pharmacokinetic/Pharmacodynamic relationships: The concentration-effect relationship over time for UACR was characterized by a maximum effect model indicating saturation at high exposures. The model-predicted time to reach the full (99%) steady-state drug effect on UACR was 138 days. The pharmacokinetic (PK) half-life was 2-3 hours and PK steady state was achieved after 2 days, indicating timescale separation.
Absorption: Finerenone is almost completely absorbed after oral administration. Absorption is rapid with maximum plasma concentrations (C
max) appearing between 0.5 and 1.25 hours after tablet intake in the fasted state. The absolute bioavailability of finerenone is 43.5% due to first-pass metabolism in the gut-wall and liver. Finerenone is not a substrate of the efflux transporter P-gp in vivo. Intake with high fat, high calorie food increased finerenone AUC by 21%, reduced C
max by 19% and prolonged the time to reach C
max to 2.5 hours. This is not clinically relevant. Therefore, finerenone can be taken with or without food (see Dosage & Administration).
Distribution: The volume of distribution at steady state (Vss) of finerenone is 52.6 L. The human plasma protein binding of finerenone in vitro is 91.7%, with serum albumin being the main binding protein.
Metabolism/Biotransformation: Approximately 90% of finerenone metabolism is mediated by CYP3A4 and 10% by CYP2C8. Four major metabolites were found in plasma, resulting from oxidation of the dihydropyridine moiety to a pyridine (M1a, M1b), subsequent hydroxylation of a methyl group (M2a) and formation of a carboxyl function (M3a). All metabolites are pharmacologically inactive.
Elimination/Excretion: The elimination of finerenone from plasma is rapid with an elimination half-life (t
1/2) of about 2 to 3 hours. Excretion of unchanged finerenone represents a minor route (<1% of dose in the urine due to glomerular filtration, <0.2% in the feces). About 80% of the administered dose was excreted via urine and approximately 20% of the dose was excreted via feces, almost exclusively in the form of metabolites. With a systemic blood clearance of about 25 L/h, finerenone can be classified as a low clearance drug.
Linearity/Non-linearity: Finerenone pharmacokinetics are linear across the investigated dose range from 1.25 to 80 mg.
Additional Information on Special Populations: Patients with renal impairment: Mild renal impairment (CLCR 60 - <90 mL/min) did not affect finerenone AUC and C
max. Compared to subjects with normal renal function (CLCR ≥90 mL/min), the effect of moderate (CLCR 30 - <60 mL/min) or severe (CLCR <30 mL/min) renal impairment on AUC of finerenone was similar with increases by 34-36%. Moderate or severe renal impairment had no effect on C
max (see Dosage & Administration).
Due to the high plasma protein binding, finerenone is not expected to be dialyzable.
Patients with hepatic impairment: There was no change in finerenone exposure in cirrhotic subjects with mild hepatic impairment (Child Pugh A) (see Dosage & Administration).
In cirrhotic subjects with moderate hepatic impairment (Child Pugh B), finerenone mean AUC was increased by 38% and C
max was unchanged compared to healthy control subjects (see Dosage & Administration).
There are no data in patients with severe hepatic impairment (Child Pugh C) (see Dosage & Administration and Precautions).
Geriatric patients: Of the 2827 patients who received Finerenone (Firialta) in the FIDELIO-DKD study, 58% of patients were 65 years and older, and 15% were 75 years and older. No overall differences in safety or efficacy were observed between these patients and younger patients.
Of the 3683 patients who received Finerenone (Firialta) in the FIGARO-DKD study, 52% of patients were 65 years and older, and 13% were 75 years and older. No overall differences in safety or efficacy were observed between these patients and younger patients.
Elderly subjects (≥65 years of age) exhibited higher finerenone plasma concentrations than younger subjects (≤45 years of age), with mean AUC and C
max values being 34% and 51% higher in the elderly (see Dosage & Administration).
Population-pharmacokinetic analyses did not identify age as a covariate for finerenone AUC or C
max.
Gender: Gender had no effect on the pharmacokinetics of finerenone (see Dosage & Administration).
Body Weight: Population-pharmacokinetic analyses identified body weight as a covariate for finerenone C
max. The C
max of a subject with a body weight of 50 kg was estimated to be 38% to 51% higher compared to a subject of 100 kg. Dose adaptation based on body weight is not warranted (see Dosage & Administration).
Ethnic differences: Population-pharmacokinetic analyses in patients demonstrated no clinically relevant difference in finerenone exposure between Asian and Caucasian patients (see Dosage & Administration).
Smoking status: Finerenone is not metabolized by an enzyme that is inducible by tobacco smoke (see Dosage & Administration).
Toxicology: Preclinical safety data: Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, single dose toxicity, genotoxicity, phototoxicity, carcinogenicity and male and female fertility.
Effects observed in repeat-dose toxicity studies were mainly due to exaggerated pharmacodynamic activities of finerenone and secondary adaptive responses.
In studies on embryo-fetal development, effects in rats were observed at exposures considered sufficiently in excess to the maximum human exposure thereby not indicating an increased concern for fetal harm. In the pre- and post-natal developmental study, adverse effects in pups exposed via milk were found. In addition, increased locomotor activity in the offspring was observed, which may result from exposure during pregnancy.
Systemic toxicity: In the animal toxicity studies, finerenone caused impaired water-electrolyte balance with a secondary response in adrenals, as expected for the mode-of-action. In the shorter-term studies in rats, additional secondary changes were found in kidneys and urinary bladder that were not reproduced in the chronic study. In addition, atrophic changes in the female genital tract of rats were found in the short-term studies at exposures representing an AUC
unbound of 19 times that in humans at a dose of 20 mg indicating little clinical relevance.
In dogs, a reduced prostate weight and size was found at an AUC
unbound of about 10 to 60 times that in humans indicating little clinical relevance.
Embryotoxicity/Teratogenicity: In the embryo-fetal toxicity in rats, finerenone resulted in reduced placental weights and signs of fetal toxicity including reduced fetal weights and retarded ossification at the maternal toxic dose of 10 mg/kg/day corresponding to an AUC
unbound of 19 times that in humans. At 30 mg/kg/ day, the incidence of visceral and skeletal variations was increased (slight edema, shortened umbilical cord, slightly enlarged fontanelle) and one fetus showed complex malformations including a rare malformation (double aortic arch) at an AUC
unbound of about 25 times that in humans. The doses free of any findings (low dose in rats, high dose in rabbits) provided safety margins of 10 to 13 times for AUC
unbound. Therefore, the findings in rats do not indicate an increased concern for fetal harm (see Use in Pregnancy & Lactation).
When rats were exposed during pregnancy and lactation in the pre- and postnatal developmental toxicity study, increased pup mortality and other adverse effects (lower pup weight, delayed pinna unfolding) were observed at about 4 times the AUC
unbound expected in humans. In addition, the offspring showed slightly increased locomotor activity, but no other neurobehavioral changes starting at about 4 times the AUC
unbound expected in humans. The dose free of findings provided a safety margin of about 2 for AUC
unbound. The increased locomotor activity in offspring may indicate a potential risk for the fetus. In addition, because of the findings in pups, a risk for the nursing infant cannot be excluded (see Precautions and Use in Pregnancy & Lactation).
Reproduction toxicity: Male fertility was not affected by Finerenone (Firialta) (see Use in Pregnancy & Lactation).
Finerenone caused reduced female fertility (decreased number of corpora lutea and implantation sites) as well as signs of early embryonic toxicity (increased post-implantational loss and decreased number of viable fetuses) at about 21 times the human AUC
unbound. In addition, reduced ovarian weights were found at about 17 times the human AUC
unbound. No effects on female fertility and early embryonic development were found at 10 times the human AUC
unbound. Therefore, the findings in female rats are of little clinical relevance (see Use in Pregnancy & Lactation).
Genotoxicity and carcinogenicity: Finerenone was non-genotoxic.
In 2-year carcinogenicity studies, finerenone did not show a carcinogenic potential in male and female rats as well as female mice. In male mice, finerenone resulted in an increase in Leydig cell adenoma at doses representing 26 times the AUC
unbound in humans. A dose representing 17 times the AUC
unbound in humans did not cause any tumors. Based on the known sensitivity of rodents to develop these tumors and the pharmacology-based mechanism at supratherapeutic doses as well as adequate safety margins, the increase in Leydig cell tumors in male mice is not clinically relevant.
Safety pharmacology: In the safety pharmacology studies assessing nervous, respiratory and cardiovascular function, the only finding was a slight shortening of the PQ interval in dogs at free plasma concentrations of about 6 times the human therapeutic concentration. Therefore, no clinical relevance is expected.
Repeated dose toxicity: In the rat 26-week study, finerenone caused slight changes in electrolytes as well as slight to moderate changes in the adrenals. These findings are related to the mode of action. Adverse effects were seen at an AUC
unbound of about 17 times that in humans (reduced body weight). The dose free of any adverse findings provided a safety margin of at least 6.
In the 4- and 13-week studies, rats showed mild degenerative changes in the kidney as well as mild changes in the urinary bladder, which were not reproduced in the chronic study. The high dose with signs of general toxicity also caused atrophic changes in female genital organs. The AUC
unbound in females at the high dose was about 21 times the human exposure. Therefore, these effects are of little clinical relevance.
In the chronic study in dogs, finerenone caused mild changes in the adrenal glands, which are regarded as mode-of-action-related. In addition, a decrease in prostate weights and size was found starting at an AUC
unbound of 10 times the maximum human therapeutic exposure. As there were not additional findings in the male genital tract at the high dose representing 60 times the maximum human exposure, this effect is of little clinical relevance.




 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image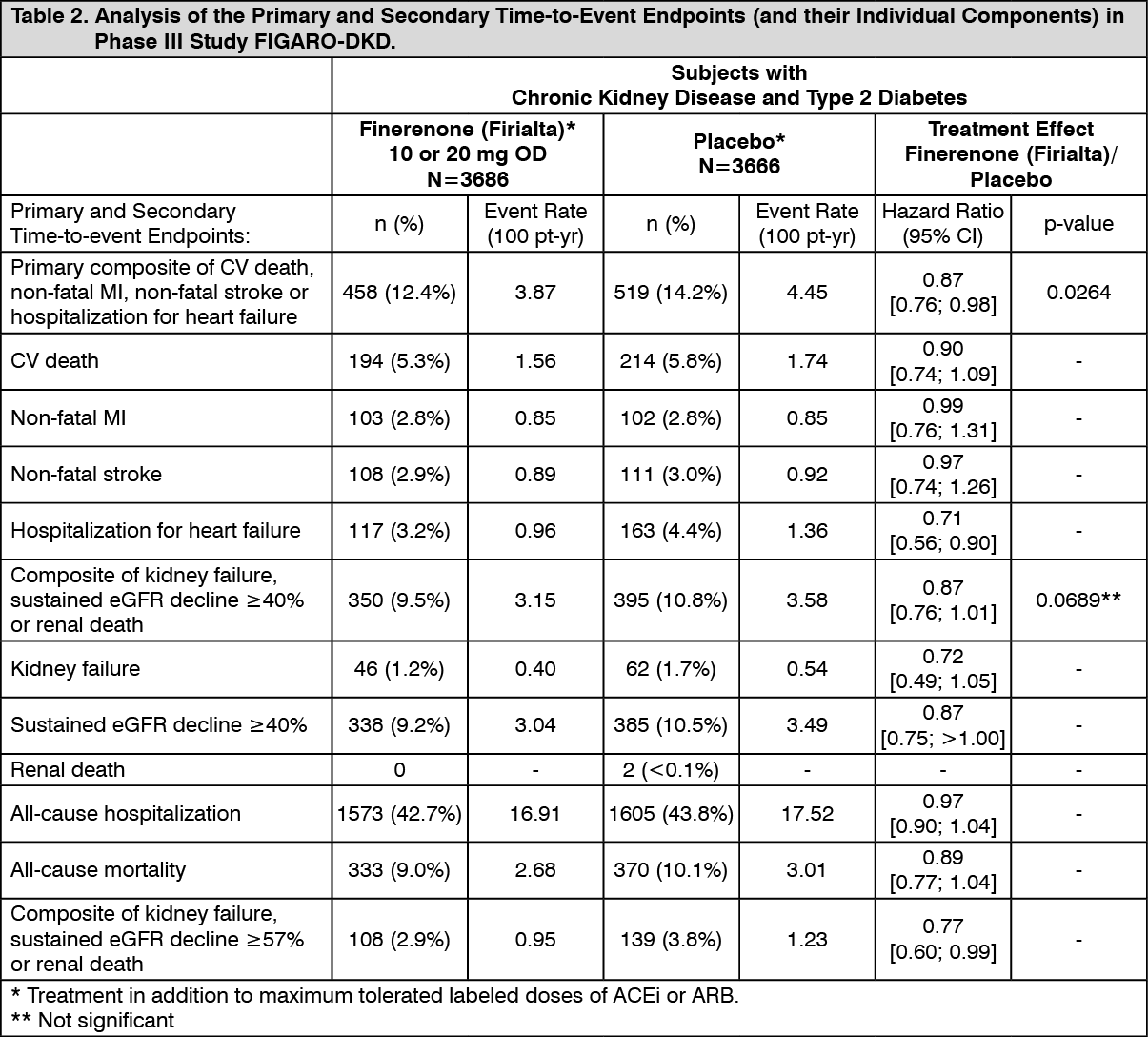 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image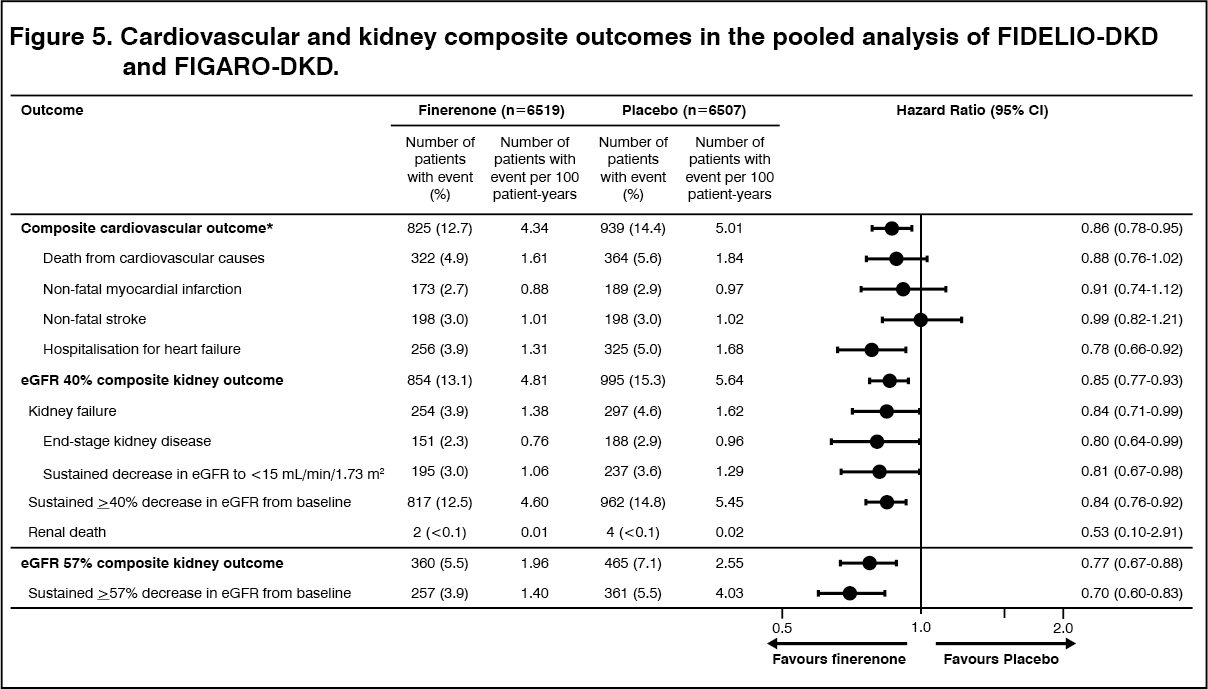 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image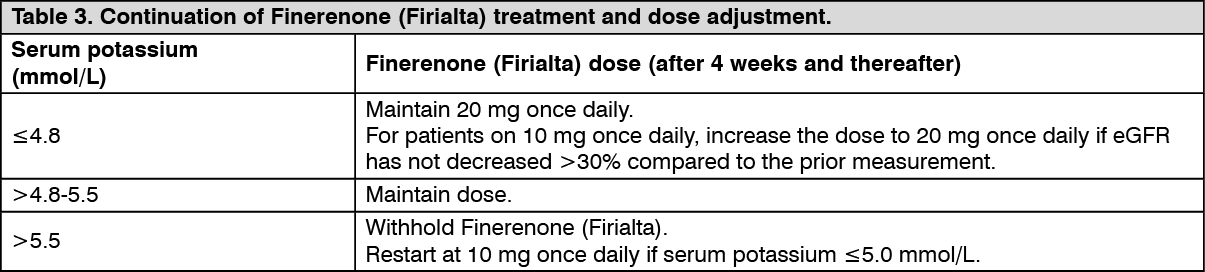 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out
