


 Sign Out
Sign Out



The adverse reactions (treatment-related adverse clinical events) most commonly observed with the use of fosphenytoin sodium (Aurantin) in clinical trials were nystagmus, dizziness, pruritus, paresthesia, somnolence, and ataxia. With two exceptions, these events are commonly associated with the administration of IV phenytoin. Paresthesia and pruritus, however, were seen much more often following fosphenytoin sodium (Aurantin) administration and occurred more often with IV fosphenytoin sodium (Aurantin) administration than with IM fosphenytoin sodium (Aurantin) administration. These events were dose and rate related; most alert patients (41 of 64; 64%) administered doses of ≥15 mg PE/kg at 150 mg PE/min experienced discomfort of some degree. Overall, sensory disturbances occurred in 13% of the patients exposed to fosphenytoin sodium (Aurantin) and were more common after IV administration, higher doses, or faster infusion rates. These sensations, generally described as itching, burning, warmth, or tingling, were usually not at the infusion site. The location of the discomfort varied, with the groin mentioned most frequently as a site of involvement. In general, these sensory disturbances occurred within several minutes of the start of the IV infusion and resolved spontaneously within 10 minutes after completion of the infusion, although some patients experienced symptoms for several hours. The sensations were not consistent with the signs of an allergic reaction and may be avoided or minimized by using a slower rate of IV infusion or by temporarily stopping the infusion. These events did not increase in severity with repeated administration and were similar to those seen with other phosphate-ester drugs such as dexamethasone phosphate.
Approximately 2% of the 873 individuals who received fosphenytoin sodium (Aurantin) in clinical trials discontinued treatment because of an adverse event. The adverse reactions most commonly associated with withdrawal were pruritus (0.7%), hypotension (0.5%), and bradycardia (0.2%).
Dose and Rate Dependency of Adverse Reactions Following IV Fosphenytoin sodium (Aurantin): The incidence of adverse reactions tended to increase as both dose and infusion rate increased. In particular, at doses of ≥15 mg PE/kg and rates ≥150 mg PE/min, transient pruritus, tinnitus, nystagmus, somnolence, and ataxia occurred 2 to 3 times more often than at lower doses or rates.
Incidence of Adverse Reactions in Aggregate Clinical Trials with IV or IM Administration: Table 1 lists the adverse reactions that occurred in at least 2% of patients treated with IV or IM fosphenytoin sodium (Aurantin); incidence in placebo is shown for comparison. Nystagmus (20.6%) and dizziness (15.6%) were the most commonly reported adverse reactions in IM or IV fosphenytoin sodium (Aurantin) patients. (See Table 1.)
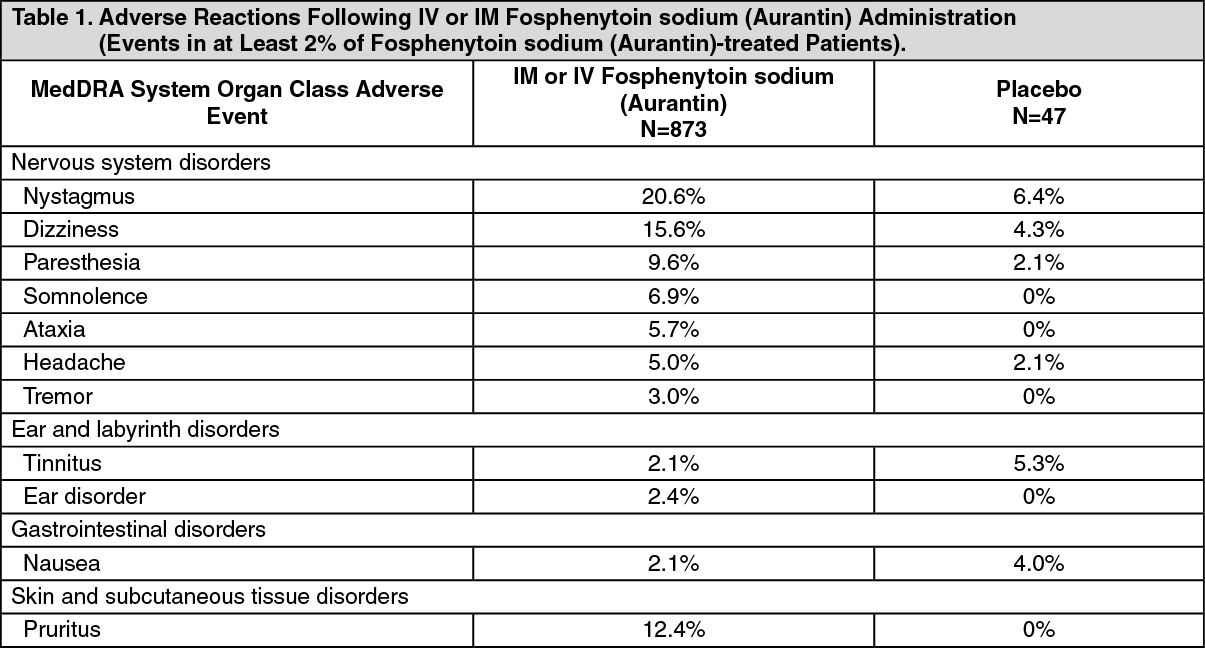 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIncidence of Adverse Reactions in a Controlled Clinical Trial: IV Administration to Adult Patients with Epilepsy or Neurosurgical Patients: Table 2 lists adverse reactions that occurred in at least 2% of adult patients treated with IV fosphenytoin sodium (Aurantin) at the maximum dose and rate in a randomized, double-blind, controlled clinical trial where the rates for phenytoin and fosphenytoin sodium (Aurantin) administration would have resulted in equivalent systemic exposure to phenytoin (Study 982-026). (See Table 2.)
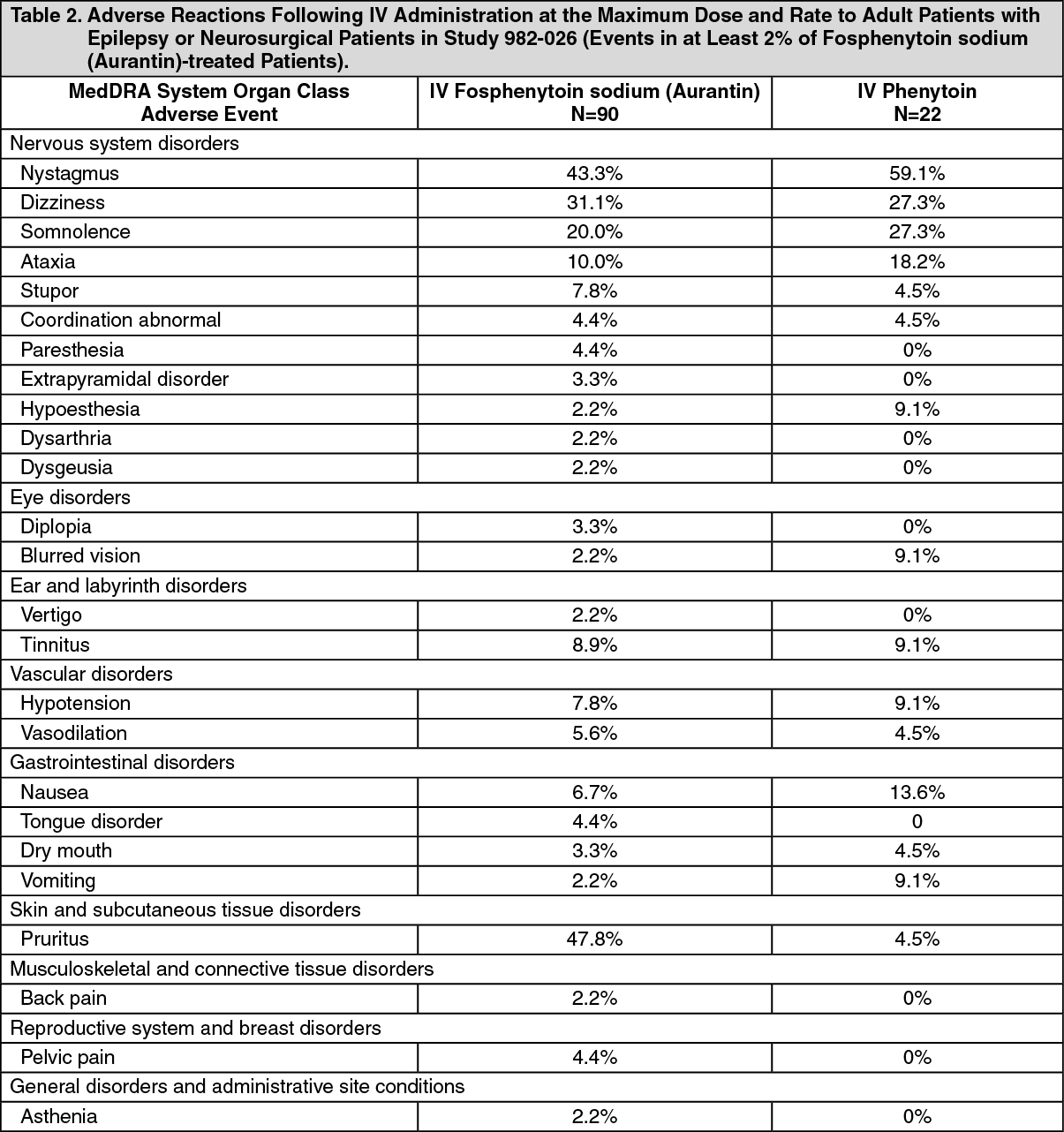 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIncidence in Controlled Clinical Trial: IV Administration to Pediatric Patients with Epilepsy or Neurosurgical Patients: The overall incidence of adverse reactions and the types of adverse reactions seen were similar among children and adults treated with fosphenytoin sodium (Aurantin). In an open-label, safety, tolerability, and pharmacokinetic study (982-028) of fosphenytoin sodium (Aurantin) in pediatric subjects (neonates through age 16), the following adverse reactions occurred at a frequency greater than 5% in 96 subjects treated with intravenous fosphenytoin sodium (Aurantin): vomiting (20.8%), nystagmus (17.7%), ataxia (10.4%), fever (8.3%), nervousness (7.3%), pruritus (6.3%), somnolence (6.3%), hypotension (5.2%), and rash (5.2%).
Incidence of Adverse Reactions in a Controlled Clinical Trial: IM Administration to Patients with Epilepsy: Table 3 lists adverse reactions that occurred in at least 2% of fosphenytoin sodium (Aurantin) treated patients in a double-blind, randomized, controlled clinical trial of adult epileptic patients receiving either IM fosphenytoin sodium (Aurantin) substituted for oral phenytoin or continuing oral phenytoin (Study 982-013). Both treatments were administered for 5 days. (See Table 3.)
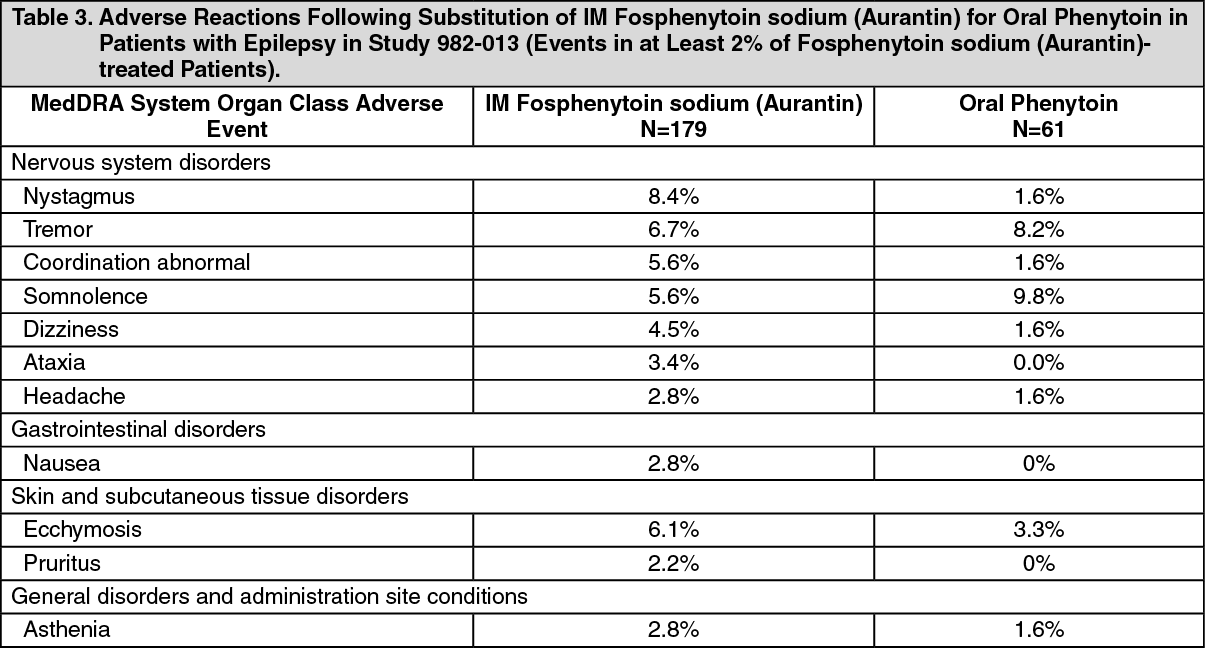 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIncidence of Adverse Reactions in a Controlled Trial: IM Administration to Patients with Epilepsy: In a double-blind study (Study 982-013) investigating temporary substitution of fosphenytoin sodium (Aurantin) for oral phenytoin, IM fosphenytoin sodium (Aurantin) was as well tolerated as IM placebo. IM fosphenytoin sodium (Aurantin) resulted in a slight increase in transient, mild to moderate itching (23% of patients vs. 11% of IM placebo-treated patients at any time during the study). This study also demonstrated that equimolar doses of IM fosphenytoin sodium (Aurantin) may be substituted for oral phenytoin sodium with no dosage adjustments needed when initiating IM therapy or returning to oral therapy. In contrast, switching between IM and oral phenytoin requires dosage adjustments because of slow and erratic phenytoin absorption from muscle.
Tolerability of Infusion: Tolerability of infusion was evaluated in clinical studies. One double-blind study (Study 982-026) assessed infusion-site tolerance of equivalent loading doses (15-20 mg PE/kg) of fosphenytoin sodium (Aurantin) infused at 150 mg PE/min or phenytoin infused at 50 mg/min. The study demonstrated better local tolerance (pain and burning at the infusion site), fewer disruptions of the infusion, and a shorter infusion period for fosphenytoin sodium (Aurantin) treated patients (see Table 4).
Fosphenytoin sodium (Aurantin)-treated patients, however, experienced more systemic sensory disturbances.
Infusion disruptions in fosphenytoin sodium (Aurantin) treated patients were primarily due to systemic burning, pruritus, and/or paresthesia while those in phenytoin-treated patients were primarily due to pain and burning at the infusion site (see Table 4).
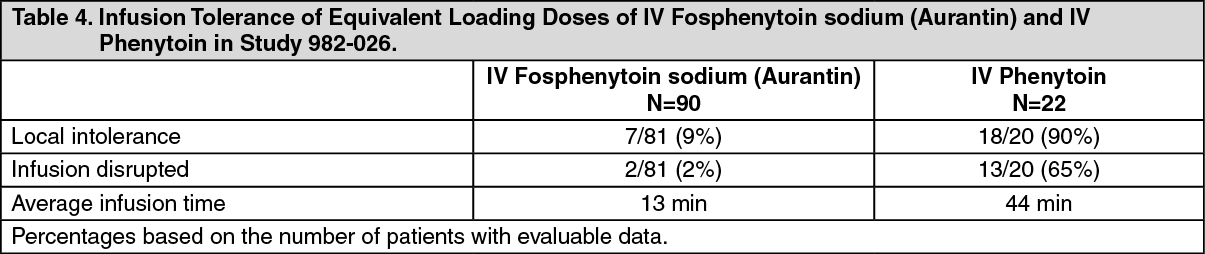 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageFrequency of Adverse Reactions: Adverse reactions in a pooled analysis of clinical trial data are listed in Table 5 by MedDRA system organ class and frequency: Very common (≥10%), common (≥1% - <10%), uncommon (≥0.1% - <1%), rare (≥0.01% - <0.1%), very rare (<0.01%). (See Table 5.)
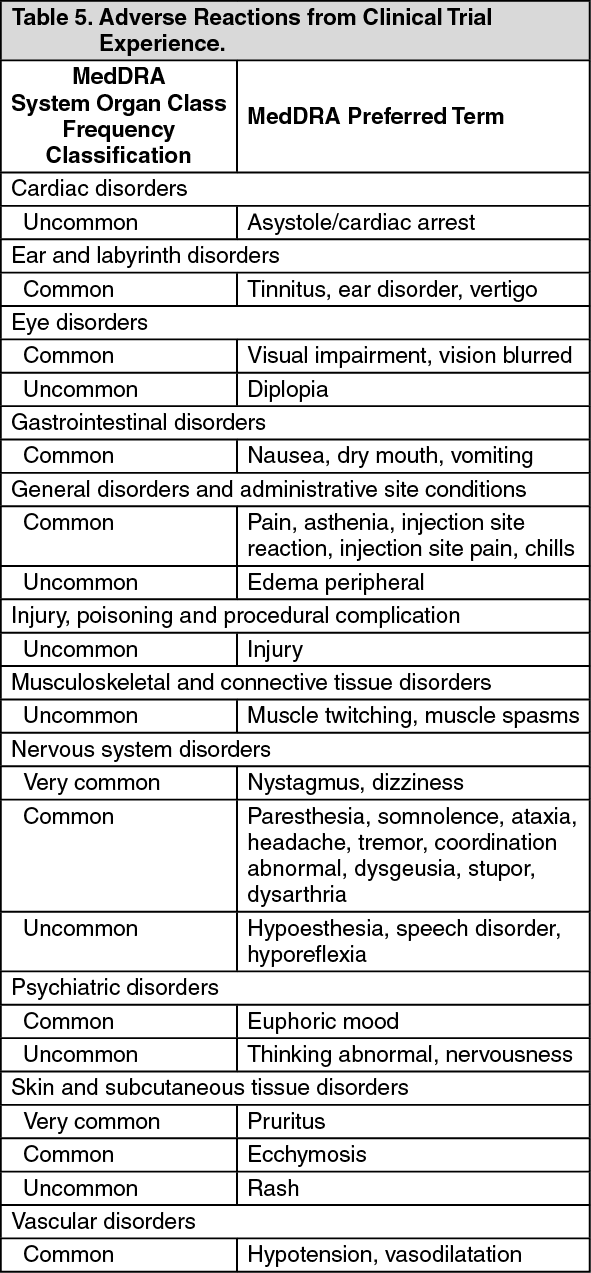 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe following adverse events (frequency unknown-cannot be estimated from available data) were reported during post-marketing surveillance: Anaphylactoid reaction, anaphylaxis, confusion, dyskinesia, Purple Glove Syndrome [see Local Toxicity (including Purple Glove Syndrome) under Precautions], and HSS/DRESS (see Hypersensitivity Syndrome Drug Reaction with Eosinophilia and Systemic Symptoms under Precautions).
View ADR Monitoring Form