


 Sign Out
Sign Out


Pharmacology: Pharmacodynamics: Mechanism of action: Atorvastatin: Atorvastatin is a selective, competitive inhibitor of HMG-CoA reductase, the rate-limiting enzyme responsible for the conversion of 3-hydroxy-3-methyl-glutaryl-coenzyme A to mevalonate, a precursor of sterols, including cholesterol. Triglycerides and cholesterol in the liver are incorporated into very low-density lipoproteins (VLDL) and released into the plasma for delivery to peripheral tissues. Low-density lipoprotein (LDL) is formed from VLDL and is catabolized primarily through the receptor with high affinity to LDL (LDL receptor).
Perindopril: Perindopril is an inhibitor of the enzyme that converts angiotensin I into angiotensin II (Angiotensin Converting Enzyme ACE). The converting enzyme, or kinase, is an exopeptidase that allows conversion of angiotensin I into the vasoconstrictor angiotensin II as well as causing the degradation of the vasodilator bradykinin into an inactive heptapeptide. Inhibition of ACE results in a reduction of angiotensin II in the plasma, which leads to increased plasma renin activity (by inhibition of the negative feedback of renin release) and reduced secretion of aldosterone. Since ACE inactivates bradykinin, inhibition of ACE also results in an increased activity of circulating and local kallikrein-kinin systems (and thus also activation of the prostaglandin system). It is possible that this mechanism contributes to the blood pressure-lowering action of ACE inhibitors and is partially responsible for certain of their side effects (e.g. cough).
Perindopril acts through its active metabolite, perindoprilat. The other metabolites show no inhibition of ACE activity in vitro.
Amlodipine: Amlodipine is a calcium ion influx inhibitor of the dihydropyridine group (slow channel blocker or calcium ion antagonist) and inhibits the transmembrane influx of calcium ions into cardiac and vascular smooth muscle.
Pharmacodynamic effects: Atorvastatin: Atorvastatin lowers plasma cholesterol and lipoprotein serum concentrations by inhibiting HMG-CoA reductase and subsequently cholesterol biosynthesis in the liver and increases the number of hepatic LDL receptors on the cell surface for enhanced uptake and catabolism of LDL.
Atorvastatin reduces LDL production and the number of LDL particles. Atorvastatin produces a profound and sustained increase in LDL receptor activity coupled with a beneficial change in the quality of circulating LDL particles. Atorvastatin is effective in reducing LDL-C in patients with homozygous familial hypercholesterolaemia, a population that has not usually responded to lipid-lowering medicinal products.
Perindopril: Hypertension: Perindopril is active in all grades of hypertension: mild, moderate, severe; a reduction in systolic and diastolic blood pressures in both supine and standing positions is observed.
Perindopril reduces peripheral vascular resistance, leading to blood pressure reduction. As a consequence, peripheral blood flow increases, with no effect on heart rate.
Renal blood flow increases as a rule, while the glomerular filtration rate (GFR) is usually unchanged.
Heart failure: Perindopril reduces cardiac work by a decrease in pre-load and after-load.
Amlodipine: The mechanism of the antihypertensive action of amlodipine is due to a direct relaxant effect on vascular smooth muscle. The precise mechanism by which amlodipine relieves angina has not been fully determined but amlodipine reduces total ischaemic burden by the following two actions: Amlodipine dilates peripheral arterioles and thus, reduces the total peripheral resistance (afterload) against which the heart works. Since the heart rate remains stable, this unloading of the heart reduces myocardial energy consumption and oxygen requirements.
The mechanism of action of amlodipine also probably involves dilatation of the main coronary arteries and coronary arterioles, both in normal and ischaemic regions. This dilatation increases myocardial oxygen delivery in patients with coronary artery spasm (Prinzmetal's or variant angina).
Clinical efficacy and safety: Triveram has not been studied on morbidity and mortality.
Atorvastatin: Atorvastatin has been shown to reduce concentrations of total-C (30% - 46%), LDL-C (41% - 61%), apolipoprotein B (34% - 50%), and triglycerides (14% - 33%) while producing variable increases in HDL-C and apolipoprotein A1 in a dose response study. These results are consistent in patients with heterozygous familial hypercholesterolaemia, nonfamilial forms of hypercholesterolaemia, and mixed hyperlipidaemia, including patients with non-insulin-dependent diabetes mellitus.
Reductions in total-C, LDL-C, and apolipoprotein B have been proven to reduce risk for cardiovascular events and cardiovascular mortality.
Homozygous familial hypercholesterolaemia: In a multicenter 8 week open-label compassionate-use study with an optional extension phase of variable length, 335 patients were enrolled, 89 of which were identified as homozygous familial hypercholesterolaemia patients. From these 89 patients, the mean percent reduction in LDL-C was approximately 20%. Atorvastatin was administered at doses up to 80 mg/day.
Prevention of cardiovascular disease: ASCOT (Anglo-Scandinavian Cardiac Outcomes Trial) is an international randomised trial with a 2x2 factorial design. ASCOT aimed to compare the effects of two antihypertensive treatment regimens in 19,257 patients (Blood Pressure Lowering Arm - ASCOT-BPLA) and the effects of the addition of atorvastatin 10 mg, compared with placebo, in 10,305 patients (Lipid Lowering Arm - ASCOT-LLA) on non-fatal and fatal coronary events.
These effects of atorvastatin on fatal and non-fatal coronary events was evaluated on hypertensive patients aged 40-79 years with no history of myocardial infarction or treatment for angina, and with TC levels ≤ 6.5 mmol/L (251 mg/dL). All patients had at least 3 of the predefined cardiovascular risk factors: male gender, age ≥55 years, smoking, diabetes, history of CHD in a first degree relative, TC:HDL C > 6, peripheral vascular disease, left ventricular hypertrophy, prior cerebrovascular event, specific ECG abnormality, proteinuria/albuminuria.
Patients received antihypertensive treatment with either amlodipine or atenolol. To achieve the goal of blood pressure control (< 140/90 mmHg in non-diabetic patients, < 130/80 mmHg in patients with diabetes), perindopril could be added in the amlodipine group and bendroflumethiazide in the atenolol group.
Patients were treated with antihypertensive therapy (either amlodipine or atenolol based regimen) and either atorvastatin 10 mg daily (n=5,168) or placebo (n=5,137).
The combination of atorvastatin and amlodipine showed a significant reduction in the primary endpoint of fatal coronary events and non-fatal myocardial infarction of 53% (95%CI [0.31; 0.69], p <0.0001) compared to placebo + amlodipine arm and of 39% (95%CI [0.08; 0.59], p <0.016) compared to atorvastatin + atenolol arm.
In a subgroup of patients from ASCOT-LLA defined in a post-hoc analysis concurrently treated with atorvastatin, perindopril and amlodipine (n=1,814), there was a 38% reduction of fatal coronary events and non-fatal myocardial infarction (95%CI [0.36; 1.08]) in comparison with atorvastatin, atenolol and bendroflumethiazide (n=1,978). There were also a significant reduction of 24% for total cardiovascular events and procedures (95%CI [0.59;0.97]), a reduction of 31% for total coronary events (95%CI [0.48;1.00]) and a significant reduction of 50% for fatal and non-fatal stroke (95%CI [0.29;0.86]), 39% for the composite of non-fatal myocardial infarction, fatal coronary events and coronary revascularization procedures (95%CI (0.38;0.97]) and 42% for the composite of cardiovascular mortality, myocardial infarction and stroke (95%CI [0.40;0.85]).
Perindopril: Hypertension: The antihypertensive activity is maximal between 4 and 6 hours after a single dose and is sustained for at least 24 hours: trough effects are about 87-100 % of peak effects.
The decrease in blood pressure occurs rapidly. In responding patients, normalisation is achieved within a month and persists without the occurrence of tachyphylaxis.
Discontinuation of treatment does not lead to a rebound effect.
Perindopril reduces left ventricular hypertrophy.
In man, perindopril has been confirmed to demonstrate vasodilatory properties. It improves large artery elasticity and decreases the media:lumen ratio of small arteries.
An adjunctive therapy with a thiazide diuretic produces an additive-type of synergy. The combination of an ACE inhibitor and a thiazide also decreases the risk of hypokalaemia induced by the diuretic treatment.
Patients with stable coronary artery disease: The EUROPA study was a multicentre, international, randomised, double-blind, placebo-controlled clinical trial lasting 4 years.
Twelve thousand two hundred and eighteen (12,218) patients aged over 18 were randomised to 8 mg perindopril tert-butylamine (equivalent to 10 mg perindopril arginine) (n=6,110) or placebo (n=6,108).
The trial population had evidence of coronary artery disease with no evidence of clinical signs of heart failure. Overall, 90% of the patients had a previous myocardial infarction and/or a previous coronary revascularisation. Most of the patients received the study medication on top of conventional therapy including platelet inhibitors, lipid lowering agents and beta-blockers.
The main efficacy criterion was the composite of cardiovascular mortality, non-fatal myocardial infarction and/or cardiac arrest with successful resuscitation. The treatment with 8 mg perindopril tert-butylamine (equivalent to 10 mg perindopril arginine) once daily resulted in a significant absolute reduction in the primary endpoint of 1.9% (relative risk reduction of 20%, 95%CI [9.4; 28.6] - p<0.001). In patients with a history of myocardial infarction and/or revascularisation, an absolute reduction of 2.2% corresponding to a RRR of 22.4% (95%CI [12.0; 31.6] - p<0.001) in the primary endpoint was observed by comparison to placebo.
Other: dual blockade of the renin-angiotensin-aldosterone system (RAAS): Two large randomised, controlled trials (ONTARGET (ONgoing Telmisartan Alone and in combination with Ramipril Global Endpoint Trial) and VA NEPHRON-D (The Veterans Affairs Nephropathy in Diabetes)) have examined the use of the combination of an ACE-inhibitor with an angiotensin II receptor blocker.
ONTARGET was a study conducted in patients with a history of cardiovascular or cerebrovascular disease, or type 2 diabetes mellitus accompanied by evidence of end-organ damage. VA NEPHRON-D was a study in patients with type 2 diabetes mellitus and diabetic nephropathy.
These studies have shown no significant beneficial effect on renal and/or cardiovascular outcomes and mortality, while an increased risk of hyperkalaemia, acute kidney injury and/or hypotension as compared to monotherapy was observed. Given their similar pharmacodynamic properties, these results are also relevant for other ACE-inhibitors and angiotensin II receptor blockers.
ACE-inhibitors and angiotensin II receptor blockers should therefore not be used concomitantly in patients with diabetic nephropathy.
ALTITUDE (Aliskiren Trial in Type 2 Diabetes Using Cardiovascular and Renal Disease Endpoints) was a study designed to test the benefit of adding aliskiren to a standard therapy of an ACE-inhibitor or an angiotensin II receptor blocker in patients with type 2 diabetes mellitus and chronic kidney disease, cardiovascular disease, or both. The study was terminated early because of an increased risk of adverse outcomes. Cardiovascular death and stroke were both numerically more frequent in the aliskiren group than in the placebo group and adverse events and serious adverse events of interest (hyperkalaemia, hypotension and renal dysfunction) were more frequently reported in the aliskiren group than in the placebo group.
Amlodipine: In patients with hypertension, once daily dosing provides clinically significant reductions of blood pressure in both the supine and standing positions throughout the 24 hour interval. Due to the slow onset of action, acute hypotension is not a feature of amlodipine administration.
In patients with angina, once daily administration of amlodipine increases total exercise time, time to angina onset, and time to 1mm ST segment depression, and decreases both angina attack frequency and glyceryl trinitrate tablet consumption.
Amlodipine has not been associated with any adverse metabolic effects or changes in plasma lipids and is suitable for use in patients with asthma, diabetes, and gout.
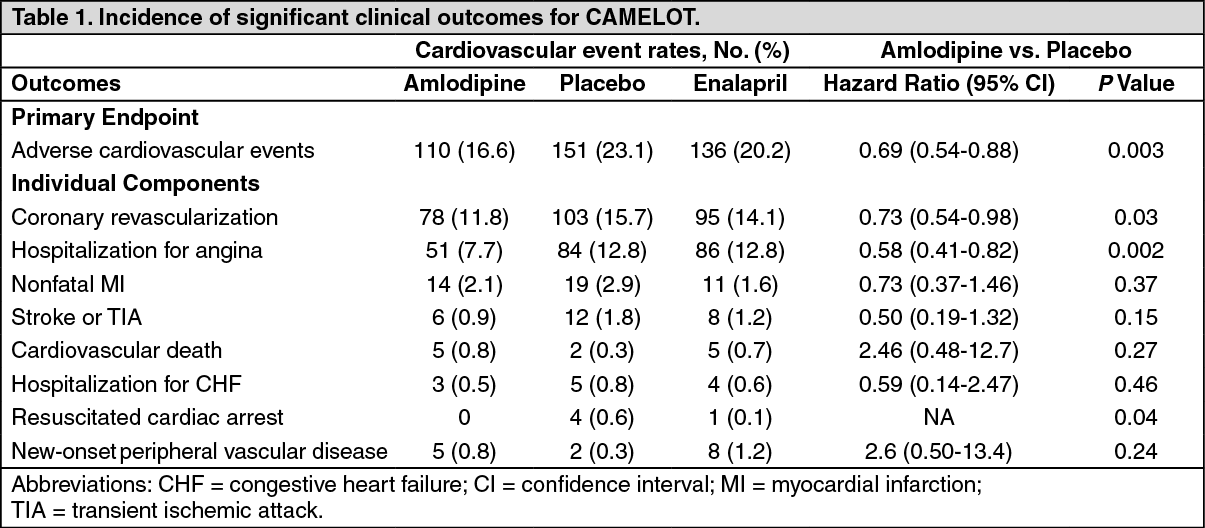
Use in patients with coronary artery disease (CAD): The effectiveness of amlodipine in preventing clinical events in patients with coronary artery disease (CAD) has been evaluated in an independent, multi-center, randomized, double- blind, placebo-controlled study of 1997 patients; Comparison of Amlodipine vs. Enalapril to Limit Occurrences of Thrombosis (CAMELOT). Of these patients, 663 were treated with amlodipine 5-10 mg, 673 patients were treated with enalapril 10-20 mg, and 655 patients were treated with placebo, in addition to standard care of statins, beta-blockers, diuretics and aspirin, for 2 years. The key efficacy results are presented in the table as follows. The results indicate that amlodipine treatment was associated with fewer hospitalizations for angina and revascularization procedures in patients with CAD. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageUse in patients with heart failure: Haemodynamic studies and exercise based controlled clinical trials in NYHA Class II-IV heart failure patients have shown that amlodipine did not lead to clinical deterioration as measured by exercise tolerance, left ventricular ejection fraction and clinical symptomatology.
A placebo controlled study (PRAISE) designed to evaluate patients in NYHA Class III-IV heart failure receiving digoxin, diuretics and ACE inhibitors has shown that amlodipine did not lead to an increase in risk of mortality or combined mortality and morbidity with heart failure.
In a follow-up, long term, placebo controlled study (PRAISE-2) of amlodipine in patients with NYHA III and IV heart failure without clinical symptoms or objective findings suggestive or underlying ischaemic disease, on stable doses of ACE inhibitors, digitalis, and diuretics, amlodipine had no effect on total cardiovascular mortality. In this same population amlodipine was associated with increased reports of pulmonary oedema.
Treatment to prevent heart attack trial (ALLHAT): A randomized double-blind morbidity-mortality study called the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT) was performed to compare newer drug therapies: amlodipine 2.5-10 mg/d (calcium channel blocker) or lisinopril 10-40 mg/d (ACE-inhibitor) as first-line therapies to that of the thiazide-diuretic, chlorthalidone 12.5-25 mg/d in mild to moderate hypertension.
A total of 33,357 hypertensive patients aged 55 or older were randomized and followed for a mean of 4.9 years. The patients had at least one additional CHD risk factor, including: previous myocardial infarction or stroke (> 6 months prior to enrolment) or documentation of other atherosclerotic CVD (overall 51.5%), type 2 diabetes (36.1%), HDL-C < 35 mg/dL (11.6%), left ventricular hypertrophy diagnosed by electrocardiogram or echocardiography (20.9%), current cigarette smoking (21.9%).
The primary endpoint was a composite of fatal CHD or non-fatal myocardial infarction. There was no significant difference in the primary endpoint between amlodipine-based therapy and chlorthalidone-based therapy: RR 0.98 95% CI (0.90-1.07) p=0.65. Among secondary endpoints, the incidence of heart failure (component of a composite combined cardiovascular endpoint) was significantly higher in the amlodipine group as compared to the chlorthalidone group (10.2% vs. 7.7%, RR 1.38, 95% CI [1.25-1.52] p<0.001). However, there was no significant difference in all-cause mortality between amlodipine-based therapy and chlorthalidone-based therapy (RR 0.96 95% CI [0.89-1.02] p=0.20).
Paediatric population: No data are available with Triveram in children.
The European Medicines Agency has granted a product-specific waiver for Triveram in all subsets of the paediatric population for the treatment of ischemic coronary artery disorders, hypertension and elevated cholesterol (see Dosage & Administration for information on paediatric use).
Pharmacokinetics: In a drug-drug interaction study in healthy subjects, co-administration of atorvastatin 40 mg, perindopril arginine 10 mg and amlodipine 10 mg resulted in a 23% increase in atorvastatin AUC, which is not clinically meaningful. The maximum concentration of perindopril was increased by about 19%, but the pharmacokinetics of perindoprilat, the active metabolite was unaffected. The rate and extent of absorption of amlodipine when co-administered with atorvastatin and perindopril were not significantly different from the rate and extent of absorption of amlodipine when taken alone.
Atorvastatin: Absorption: Atorvastatin is rapidly absorbed after oral administration; maximum plasma concentrations (Cmax) occur within 1 to 2 hours. Extent of absorption increases in proportion to atorvastatin dose. After oral administration, atorvastatin film-coated tablets are 95% to 99% bioavailable compared to the oral solution. The absolute bioavailability of atorvastatin is approximately 12% and the systemic availability of HMG-CoA reductase inhibitory activity is approximately 30%. The low systemic availability is attributed to presystemic clearance in gastrointestinal mucosa and/or hepatic first-pass metabolism.
Distribution: Mean volume of distribution of atorvastatin is approximately 381 l. Atorvastatin is ≥ 98% bound to plasma proteins.
Biotransformation: Atorvastatin is metabolized by cytochrome P450 3A4 to ortho- and parahydroxylated derivatives and various beta-oxidation products. Apart from other pathways these products are further metabolized via glucuronidation. In vitro, inhibition of HMG-CoA reductase by ortho- and parahydroxylated metabolites is equivalent to that of atorvastatin. Approximately 70% of circulating inhibitory activity for HMG-CoA reductase is attributed to active metabolites.
Elimination: Atorvastatin is eliminated primarily in bile following hepatic and/or extrahepatic metabolism. However, atorvastatin does not appear to undergo significant enterohepatic recirculation. Mean plasma elimination half-life of atorvastatin in humans is approximately 14 hours. The half-life of inhibitory activity for HMG-CoA reductase is approximately 20 to 30 hours due to the contribution of active metabolites.
Atorvastatin is a substrate of the hepatic transporters, organic anion-transporting polypeptide 1B1 (OATP1B1) and 1B3 (OATP1B3) transporter. Metabolites of atorvastatin are substrates of OATP1B1. Atorvastatin is also identified as a substrate of the efflux transporters multi-drug resistance protein 1 (MDR1) and breast cancer resistance protein (BCRP), which may limit the intestinal absorption and biliary clearance of atorvastatin.
Special populations: Elderly: Plasma concentrations of atorvastatin and its active metabolites are higher in healthy elderly subjects than in young adults while the lipid effects were comparable to those seen in younger patient populations.
Gender: Concentrations of atorvastatin and its active metabolites in women differ from those in men (Women: approx. 20% higher for Cmax and approx. 10% lower for AUC). These differences were of no clinical significance, resulting in no clinically significant differences in lipid effects among men and women.
Renal impairment: Renal disease has no influence on the plasma concentrations or lipid effects of atorvastatin and its active metabolites.
Hepatic impairment: Plasma concentrations of atorvastatin and its active metabolites are markedly increased (approx. 16-fold in Cmax and approx. 11-fold in AUC) in patients with chronic alcoholic liver disease (Child-Pugh B).
SLOC1B1 polymorphism: Hepatic uptake of all HMG-CoA reductase inhibitors including atorvastatin, involves the OATP1B1 transporter. In patients with SLCO1B1 polymorphism there is a risk of increased exposure of atorvastatin, which may lead to an increased risk of rhabdomyolysis (see Precautions).
Polymorphism in the gene encoding OATP1B1 (SLCO1B1 c.521CC) is associated with a 2.4-fold higher atorvastatin exposure (AUC) than in individuals without this genotype variant (c.521TT). A genetically impaired hepatic uptake of atorvastatin is also possible in these patients. Possible consequences for the efficacy are unknown.
Perindopril: Absorption: After oral administration, the absorption of perindopril is rapid and the peak concentration is achieved within 1 hour. The plasma half-life of perindopril is equal to 1 hour.
Biotransformation: Perindopril is a prodrug. Twenty seven percent of the administered perindopril dose reaches the bloodstream as the active metabolite perindoprilat. In addition to active perindoprilat, perindopril yields five metabolites, all inactive. The peak plasma concentration of perindoprilat is achieved within 3 to 4 hours.
As ingestion of food decreases conversion to perindoprilat, hence bioavailability, perindopril arginine should be administered orally in a single daily dose in the morning before a meal.
Linearity: It has been demonstrated a linear relationship between the dose of perindopril and its plasma exposure.
Distribution: The volume of distribution is approximately 0.2 L/kg for unbound perindoprilat. Protein binding of perindoprilat to plasma proteins is 20%, principally to angiotensin converting enzyme, but is concentration-dependent.
Elimination: Perindoprilat is eliminated in the urine and the terminal half-life of the unbound fraction is approximately 17 hours, resulting in steady-state within 4 days.
Special populations: Elderly: Elimination of perindoprilat is decreased in elderly, and also in patients with heart or renal failure.
Renal impairment: Dosage adjustment in renal insufficiency is desirable depending on the degree of impairment (creatinine clearance).
Dialysis clearance of perindoprilat is equal to 70 mL/min.
In patients with cirrhosis: Perindopril kinetics are modified in patients with cirrhosis: hepatic clearance of the parent molecule is reduced by half. However, the quantity of perindoprilat formed is not reduced and therefore no dosage adjustment is required (see Dosage & Administration and Precautions).
Amlodipine: Absorption: After oral administration of therapeutic doses, amlodipine is well absorbed with peak blood levels between 6-12 hours post dose. Absolute bioavailability has been estimated to be between 64 and 80%. The bioavailability of amlodipine is not affected by food intake.
Distribution: The volume of distribution is approximately 21 L/kg. In vitro studies have shown that approximately 97.5% of circulating amlodipine is bound to plasma proteins.
Biotransformation and elimination: The terminal plasma elimination half life is about 35-50 hours and is consistent with once daily dosing. Amlodipine is extensively metabolised by the liver to inactive metabolites with 10% of the parent compound and 60% of metabolites excreted in the urine.
Special populations: Hepatic impairment: Very limited clinical data are available regarding amlodipine administration in patients with hepatic impairment. Patients with hepatic insufficiency have decreased clearance of amlodipine resulting in a longer half-life and an increase in AUC of approximately 40-60%.
Elderly: The time to reach peak plasma concentrations of amlodipine is similar in elderly and younger subjects. Amlodipine clearance tends to be decreased with resulting increases in AUC and elimination half-life in elderly patients. Increases in AUC and elimination half-life in patients with congestive heart failure were as expected for the patient age group studied.
Toxicology: Preclinical safety data: No preclinical studies have been performed with Triveram.
Atorvastatin: Reproductive toxicology and effect on fertility: There is evidence from animal experimental studies that HMG-CoA reductase inhibitors may affect the development of embryos or fetuses. In rats, rabbits and dogs atorvastatin had no effect on fertility and was not teratogenic. However, at maternally toxic doses, fetal toxicity was observed in rats and rabbits. The development of the rat offspring was delayed and post-natal survival reduced during exposure of the dams to high doses of atorvastatin. In rats, there is evidence of placental transfer. In rats, plasma concentrations of atorvastatin are similar to those in milk. It is not known whether atorvastatin or its metabolites are excreted in human milk.
Carcinogenesis, mutagenesis: Atorvastatin was negative for mutagenic and clastogenic potential in a battery of 4 in vitro tests and 1 in vivo assay. Atorvastatin was not found to be carcinogenic in rats, but high doses in mice (resulting in 6-11 fold the AUC0-24h reached in humans at the highest recommended dose) showed hepatocellular adenomas in males and hepatocellular carcinomas in females.
Perindopril: Chronic toxicity: In the chronic oral toxicity studies (rats and monkeys), the target organ is the kidney, with reversible damage.
Reproductive toxicology and effect on fertility: Reproduction toxicology studies (rats, mice, rabbits and monkeys) showed no sign of embryotoxicity or teratogenicity. However, angiotensin converting enzyme inhibitors, as a class, have been shown to induce adverse effects on late fetal development, resulting in fetal death and congenital effects in rodents and rabbits: renal lesions and an increase in peri- and postnatal mortality have been observed. Fertility was not impaired either in male or in female rats.
Carcinogenesis, mutagenesis: No mutagenicity has been observed in in vitro or in vivo studies. No carcinogenicity has been observed in long term studies in rats and mice.
Amlodipine: Reproductive toxicology: Reproductive studies in rats and mice have shown delayed date of delivery, prolonged duration of labour and decreased pup survival at dosages approximately 50 times greater than the maximum recommended dosage for humans based on mg/kg.
Impairment of fertility: There was no effect on the fertility of rats treated with amlodipine (males for 64 days and females 14 days prior to mating) at doses up to 10 mg/kg/day (8 times* the maximum recommended human dose of 10 mg on a mg/m2 basis). In another rat study in which male rats were treated with amlodipine besylate for 30 days at a dose comparable with the human dose based on mg/kg, decreased plasma follicle-stimulating hormone and testosterone were found as well as decreases in sperm density and in the number of mature spermatids and Sertoli cells.
Carcinogenesis, mutagenesis: Rats and mice treated with amlodipine in the diet for two years, at concentrations calculated to provide daily dosage levels of 0.5, 1.25, and 2.5 mg/kg/day showed no evidence of carcinogenicity. The highest dose (for mice, similar to, and for rats twice* the maximum recommended clinical dose of 10 mg on a mg/m2 basis) was close to the maximum tolerated dose for mice but not for rats.
Mutagenicity studies revealed no drug related effects at either the gene or chromosome levels.
*Based on patient weight of 50 kg.