


 Sign Out
Sign Out


Pharmacology: Pharmacodynamics: Mechanism of action: Abiraterone acetate (ZYTIGA) is converted in vivo to abiraterone, an androgen biosynthesis inhibitor. Specifically, abiraterone selectively inhibits the enzyme 17α-hydroxylase/C17,20-lyase (CYP17). This enzyme is expressed in and is required for androgen biosynthesis in testicular, adrenal and prostatic tumour tissues. CYP17 catalyses the conversion of pregnenolone and progesterone into testosterone precursors, DHEA and androstenedione, respectively, by 17α-hydroxylation and cleavage of the C17,20 bond. CYP17 inhibition also results in increased mineralocorticoid production by the adrenals (see Precautions).
Androgen-sensitive prostatic carcinoma responds to treatment that decreases androgen levels. Androgen deprivation therapies, such as treatment with LHRH agonists or orchiectomy, decrease androgen production in the testes but do not affect androgen production by the adrenals or in the tumor. Treatment with ZYTIGA decreases serum testosterone to undetectable levels (using commercial assays) when given with LHRH agonists (or orchiectomy).
Pharmacodynamic effects: ZYTIGA decreases serum testosterone and other androgens to levels lower than those achieved by the use of LHRH agonists alone or by orchiectomy. This results from the selective inhibition of the CYP17 enzyme required for androgen biosynthesis. Prostate specific antigen (PSA) serves as a biomarker in patients with prostate cancer. In a Phase 3 clinical study of patients who failed prior chemotherapy with taxanes, 38% of patients treated with ZYTIGA, versus 10% of patients treated with placebo, had at least a 50% decline from baseline in PSA levels.
Use of Spironolactone: Patients in pivotal clinical trials with ZYTIGA were not allowed to use spironolactone as spironolactone binds to the androgen receptor and may increase PSA levels.
Clinical studies: The efficacy of ZYTIGA was established in three randomized placebo-controlled multicenter Phase 3 clinical studies (Studies 3011, 302, and 301) of patients with mHSPC and mCRPC.
Study 3011 enrolled patients who were newly diagnosed (within 3 months of randomization) mHSPC who had high-risk prognostic factors. High-risk prognosis was defined as having at least 2 of the following 3 risk factors: (1) Gleason score of ≥8; (2) presence of 3 or more lesions on bone scan; (3) presence of measurable visceral (excluding lymph node disease) metastasis. In the active arm, ZYTIGA was administered at a dose of 1000 mg daily in combination with low dose prednisone or prednisolone 5 mg once daily in addition to ADT (LHRH agonist or orchiectomy), which was the standard of care treatment. Patients in the control arm received ADT and placebos for both ZYTIGA and prednisone.
Study 302 enrolled patients who were asymptomatic or mildly symptomatic and had not received prior chemotherapy, whereas Study 301 enrolled patients who received prior chemotherapy containing a taxane. In both studies patients were using an LHRH agonist or were previously treated with orchiectomy. In the active treatment arms, ZYTIGA was administered at a dose of 1000 mg daily in combination with low dose prednisone or prednisolone 5 mg twice daily. Control patients received placebo and low dose prednisone or prednisolone 5 mg twice daily.
Because changes in PSA serum concentration do not always predict clinical benefit, in all studies patients were maintained on ZYTIGA until discontinuation criteria were met as specified for each study as follows.
Study 3011 (patients with newly diagnosed high risk mHSPC): In Study 3011, (n=1199) the median age of enrolled patients was 67 years. The number of patients treated with ZYTIGA by racial group was Caucasian 832 (69.4%), Asian 246 (20.5%), Black or African American 25 (2.1%), other 80 (6.7%), unknown/not reported 13 (1.1%), and American Indian or Alaska Native 3 (0.3%). The ECOG performance status was 0 or 1 for 97% of patients. Patients with known brain metastasis, uncontrolled hypertension, significant heart disease, or NYHA Class II-IV heart failure were excluded. Patients that were treated with prior pharmacotherapy, radiation therapy, or surgery for metastatic prostate cancer were excluded with the exception of up to 3 months of ADT or 1 course of palliative radiation or surgical therapy to treat symptoms resulting from metastatic disease. Co-primary efficacy endpoints were overall survival (OS) and radiographic progression-free survival (rPFS). The median baseline pain score, as measured by the Brief Pain Inventory Short Form (BPI-SF) was 2.0 in both the treatment and Placebo groups. In addition to the co-primary endpoint measures, benefit was also assessed using time to skeletal-related event (SRE), time to subsequent therapy for prostate cancer, time to initiation of chemotherapy, time to pain progression, and time to PSA progression. Treatment continued until disease progression, withdrawal of consent, the occurrence of unacceptable toxicity, or death.
Radiographic progression-free survival was defined as the time from randomisation to the occurrence of radiographic progression or death from any cause. Radiographic progression included progression by bone scan (according to modified PCWG2) or progression of soft tissue lesions by CT or MRI (according to RECIST 1.1).
A significant difference in rPFS between treatment groups was observed (see Table 1 and Figure 1).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image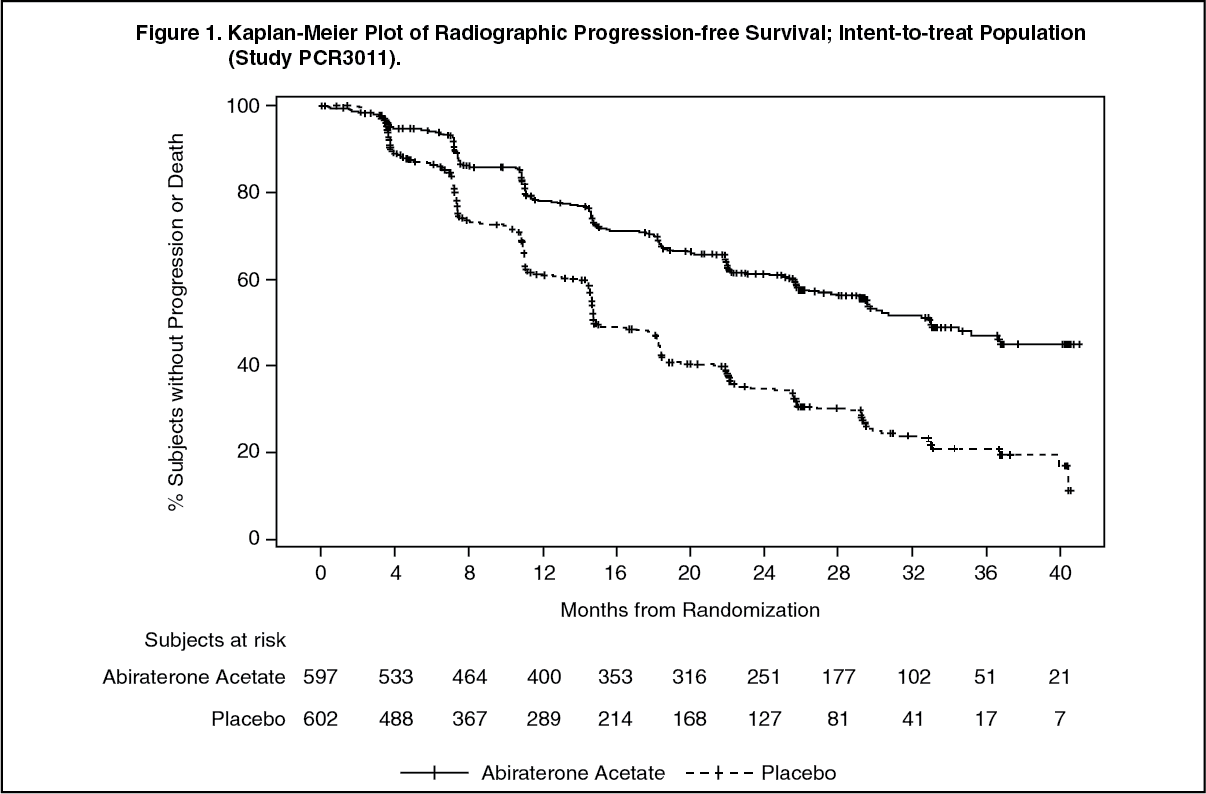 Click on icon to see table/diagram/image
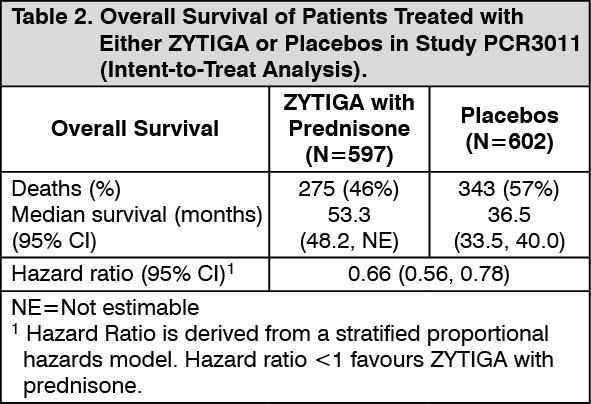
Click on icon to see table/diagram/imageA statistically significant improvement in OS in favour of AA-P plus ADT was observed with a 38% reduction in the risk of death compared to Placebo plus ADT (HR=0.621; 95% CI: 0.509, 0.756; p<0.0001), crossing the pre-specified boundary for OS at Interim Analysis 1 of 0.010 (see Table 2 and Figure 2).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image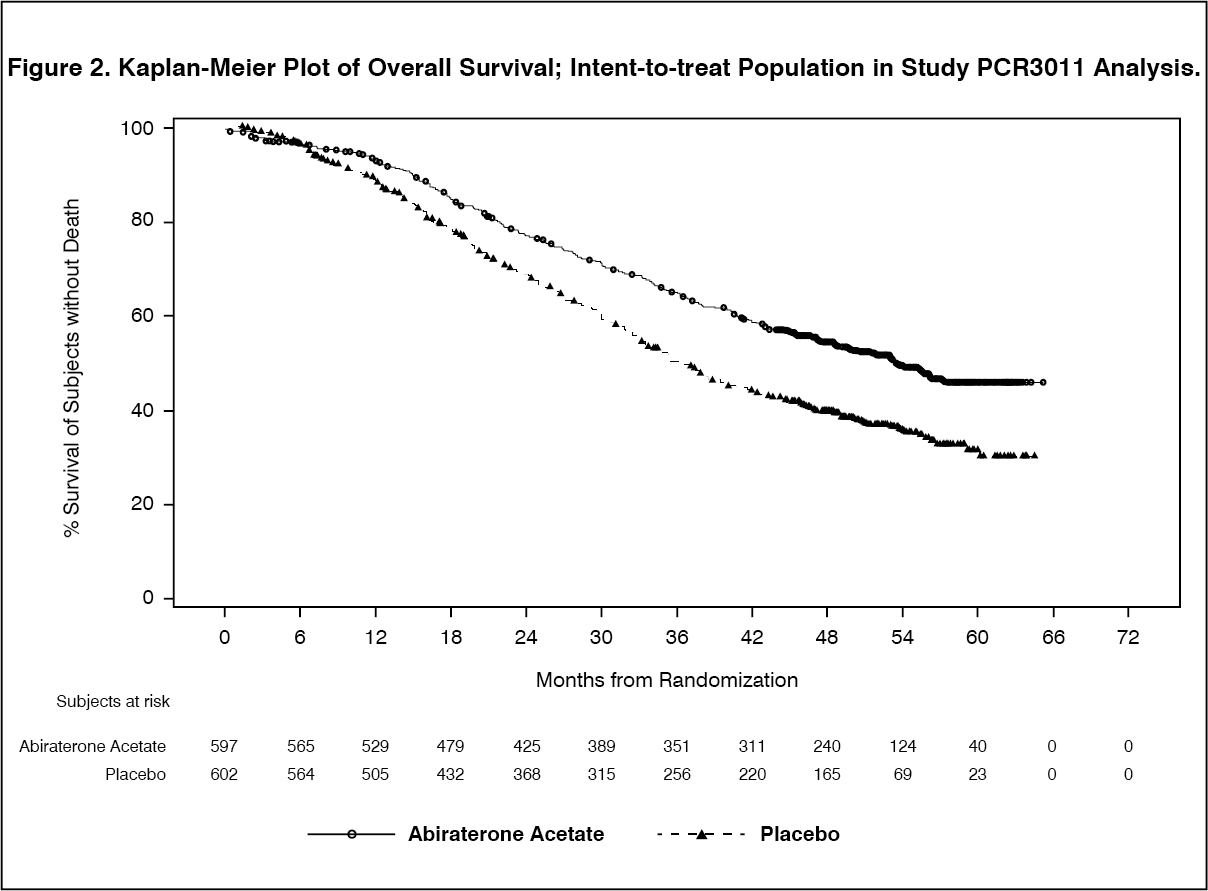 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSubgroup analyses consistently favour treatment with ZYTIGA. The treatment effect of AA-P on rPFS and OS across the pre-specified subgroups was favourable and consistent with the overall study population, except for the subgroup of ECOG score of 2 where no trend towards benefit was observed, however the small sample size (n=40) limits drawing any meaningful conclusion.
In addition to the observed improvements in overall survival and rPFS, benefit was demonstrated for ZYTIGA vs. placebo treatment in all prospectively-defined secondary endpoints.
Study 302 (asymptomatic or mildly symptomatic patients who did not receive prior chemotherapy): In Study 302, (n=1088) the median age of enrolled patients was 71 years for patients treated with ZYTIGA plus prednisone or prednisolone and 70 years for patients treated with placebo plus prednisone or prednisolone. The ECOG performance status was 0 for 76% of patients, and 1 for 24% of patients in both arms. Patients with visceral metastases were excluded. Co-primary efficacy endpoints were overall survival and radiographic progression-free survival (rPFS). Baseline pain assessment was 0-1 (asymptomatic) in 66% of patients and 2-3 (mildly symptomatic) in 26% of patients as defined by the Brief Pain Inventory-Short Form (worst pain over the last 24 hours). In addition to the co-primary endpoint measures, benefit was also assessed using time to opiate use for cancer pain, time to initiation of cytotoxic chemotherapy, time to deterioration in ECOG performance score by ≥1 point and time to PSA progression based on Prostate Cancer Working Group-2 (PCWG2) criteria.
In the 302 study treatments were discontinued at the time of unequivocal clinical progression. Treatments could also be discontinued at the time of confirmed radiographic progression at the discretion of the investigator.
Radiographic progression free survival was assessed with the use of sequential imaging studies as defined by PCWG2 criteria (for bone lesions) and modified Response Evaluation Criteria In Solid Tumours (RECIST) criteria (for soft tissue lesions). Analysis of rPFS utilized centrally-reviewed radiographic assessment of progression.
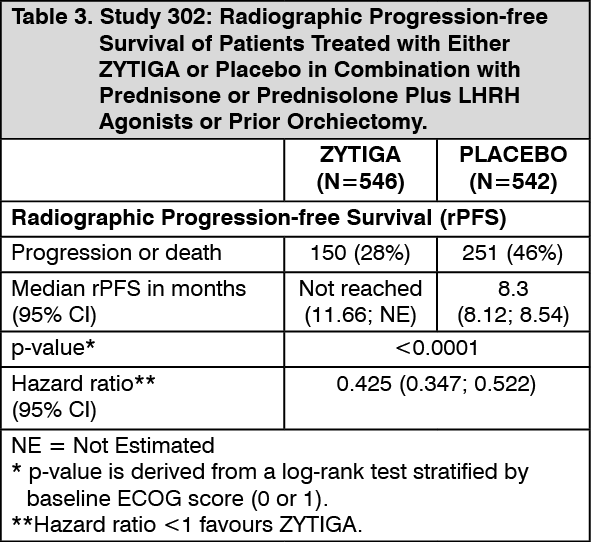
At the planned rPFS analysis there were 401 events; 150 (28%) of patients treated with ZYTIGA and 251 (46%) of patients treated with placebo had radiographic evidence of progression or had died. A significant difference in rPFS between treatment groups was observed (see Table 3 and Figure 3).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image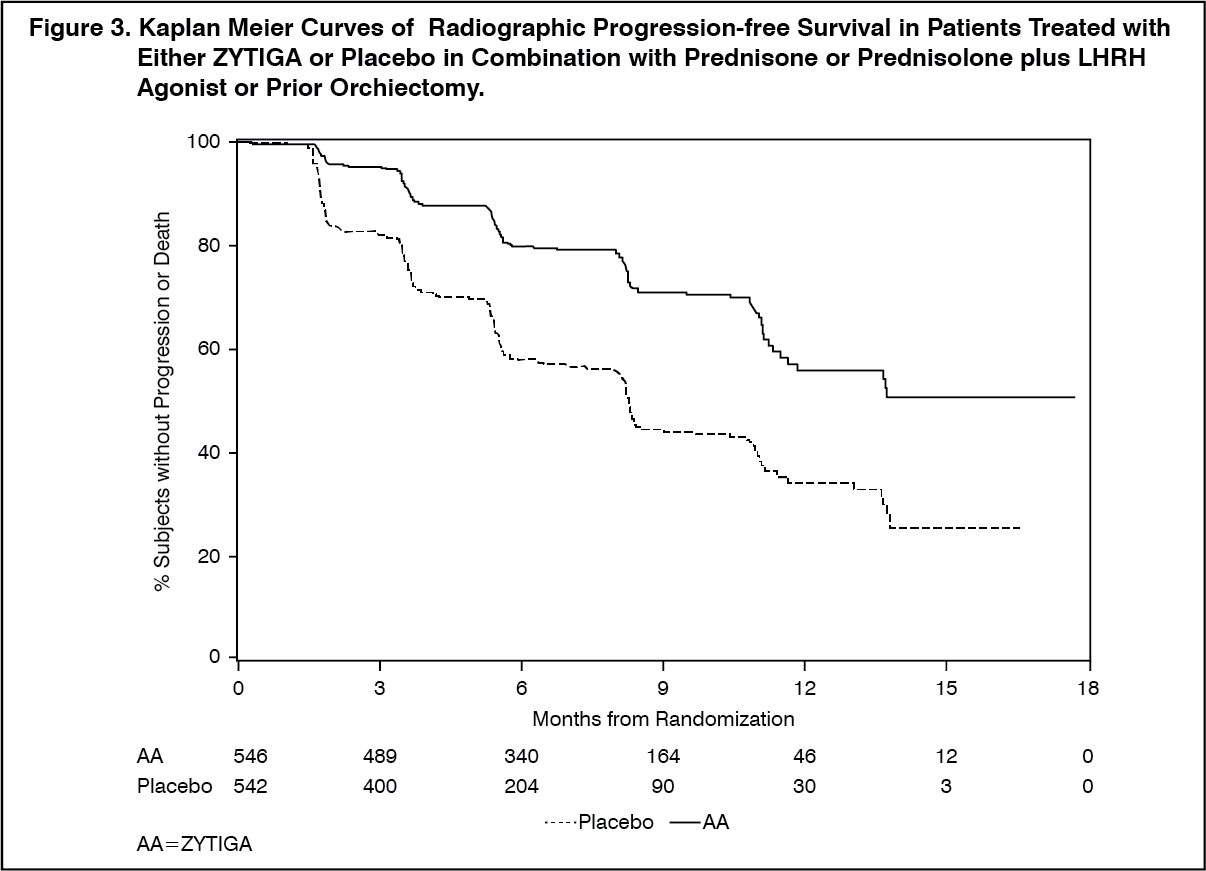 Click on icon to see table/diagram/image
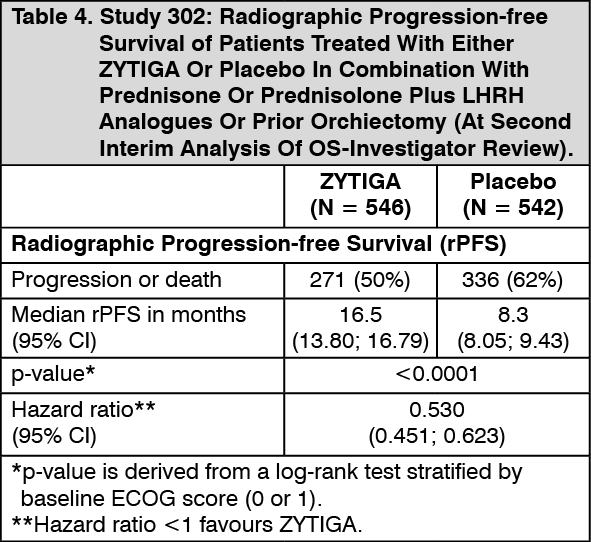
Click on icon to see table/diagram/imageHowever, subject data continued to be collected through the date of the second interim analysis of Overall survival (OS). The investigator radiographic review of rPFS performed as a follow up sensitivity analysis is presented in Table 4 and Figure 4.
Six hundred and seven (607) subjects had radiographic progression or died: 271 (50%) in the abiraterone acetate group and 336 (62%) in the placebo group. Treatment with abiraterone acetate decreased the risk of radiographic progression or death by 47% compared with placebo (HR = 0.530; 95% CI: [0.451, 0.623], p < 0.0001). The median rPFS was 16.5 months in the abiraterone acetate group and 8.3 months in the placebo group. (See Table 4 and Figure 4).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image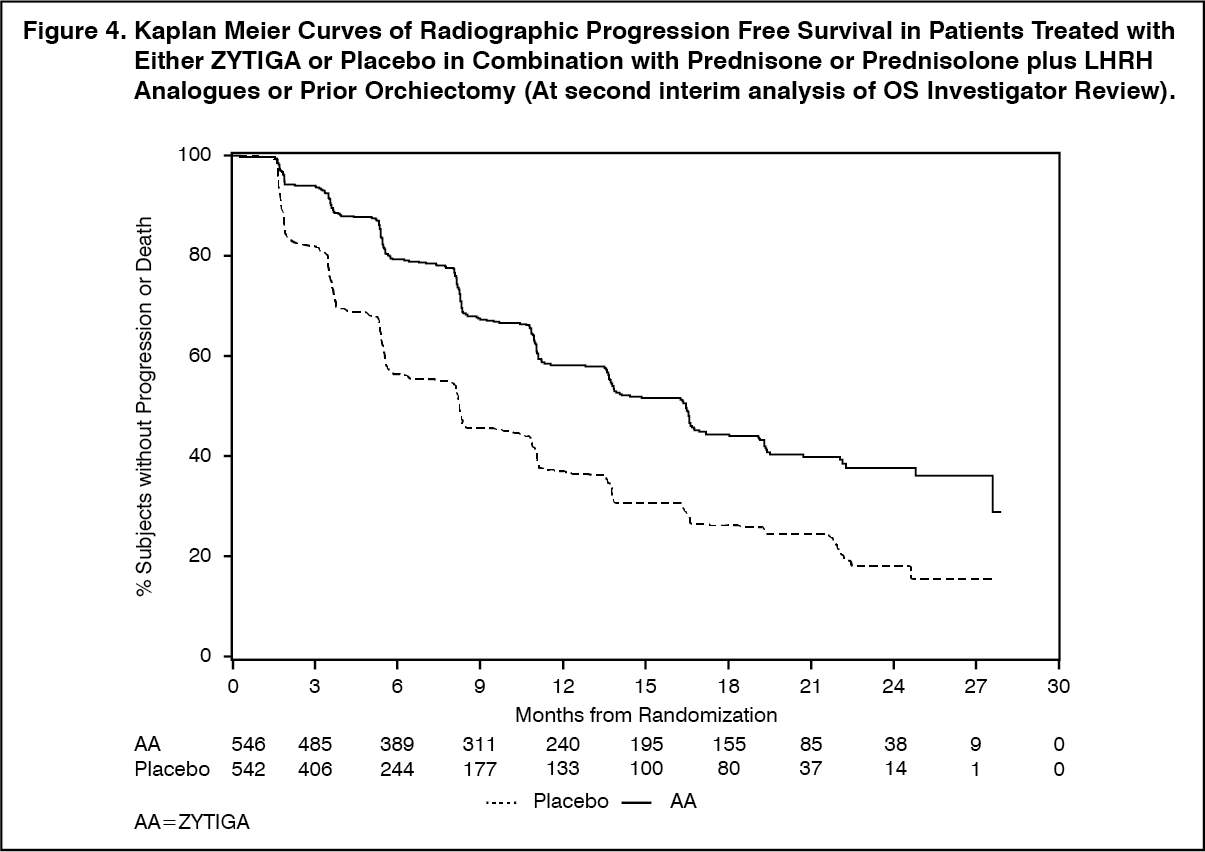 Click on icon to see table/diagram/image
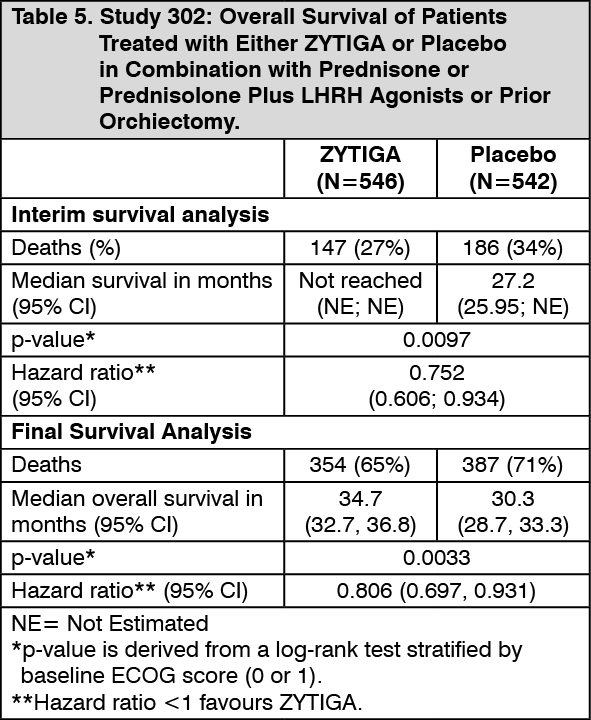
Click on icon to see table/diagram/imageA planned interim analysis (IA) for overall survival was conducted after 333 deaths were observed. The study was unblinded based on the magnitude of clinical benefit observed and patients in the placebo group were offered treatment with ZYTIGA. Overall survival was longer for ZYTIGA than placebo with a 25% reduction in risk of death (HR = 0.752; 95 % CI: [0.606, 0.934], p = 0.0097), but OS was not mature and interim results did not meet the pre-specified stopping boundary for statistical significance (see Table 5). Survival continued to be followed after this IA.
The planned final analysis for OS was conducted after 741 deaths were observed (median follow-up of 49 months). Sixty five percent (354 of 546) of patients treated with ZYTIGA, compared with 71% (387 of 542) of patients treated with placebo, had died. A statistically significant OS benefit in favor of the ZYTIGA-treated group was demonstrated with a 19.4% reduction in risk of death (HR = 0.806; 95% CI: [0.697, 0.931], p = 0.0033) and an improvement in median OS of 4.4 months (ZYTIGA 34.7 months, placebo 30.3 months) (see Table 5 and Figure 5). This improvement was demonstrated despite subsequent therapy being common, irrespective of whether patients initially received abiraterone acetate or placebo. Subsequent therapies in the abiraterone acetate and placebo patient groups included abiraterone acetate, 69 (13%) and 238 (44%); docetaxel, 311 (57%) and 331 (61%); cabazitaxel, 100 (18%) and 105 (19%); and enzalutamide 87 (16%) and 54 (10%) patients respectively. (See Table 5 and Figure 5).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image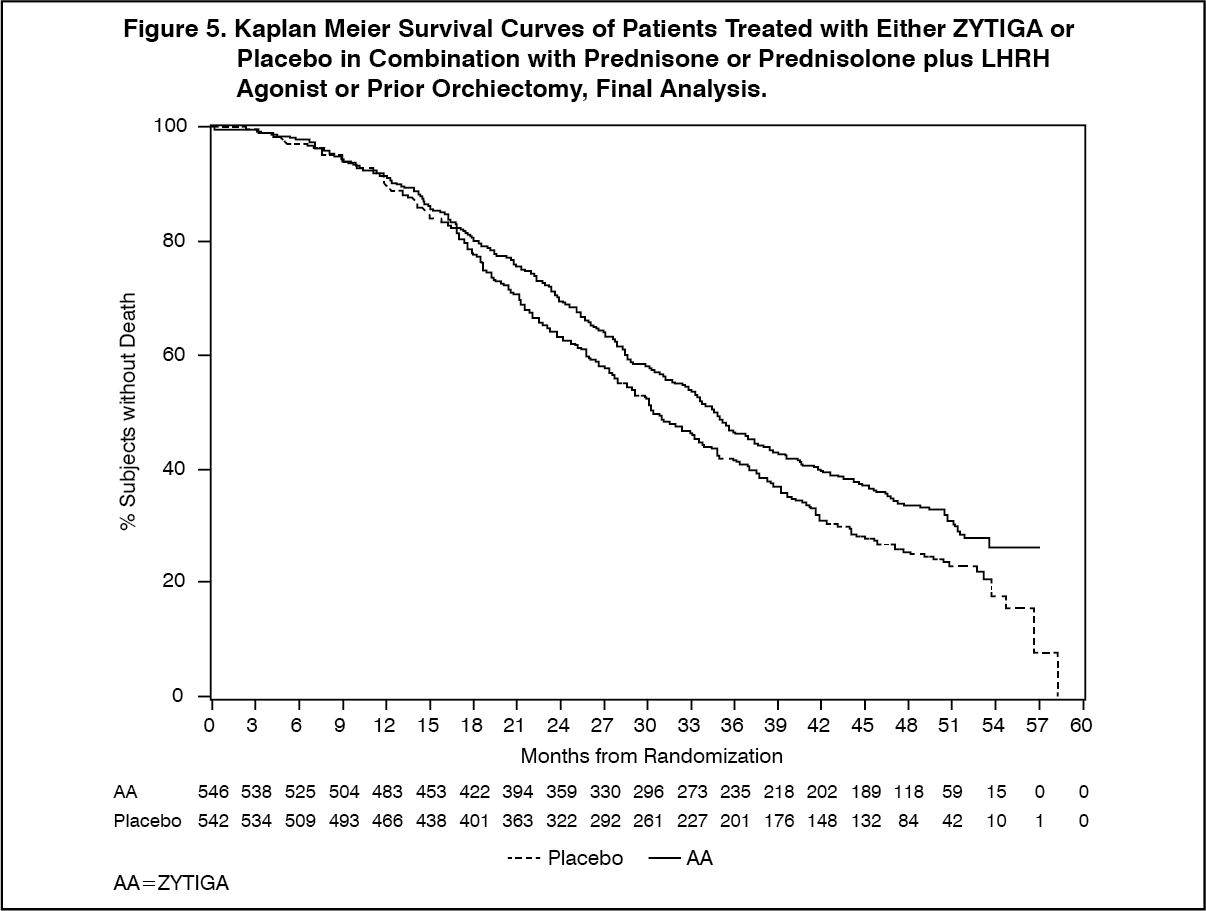 Click on icon to see table/diagram/image
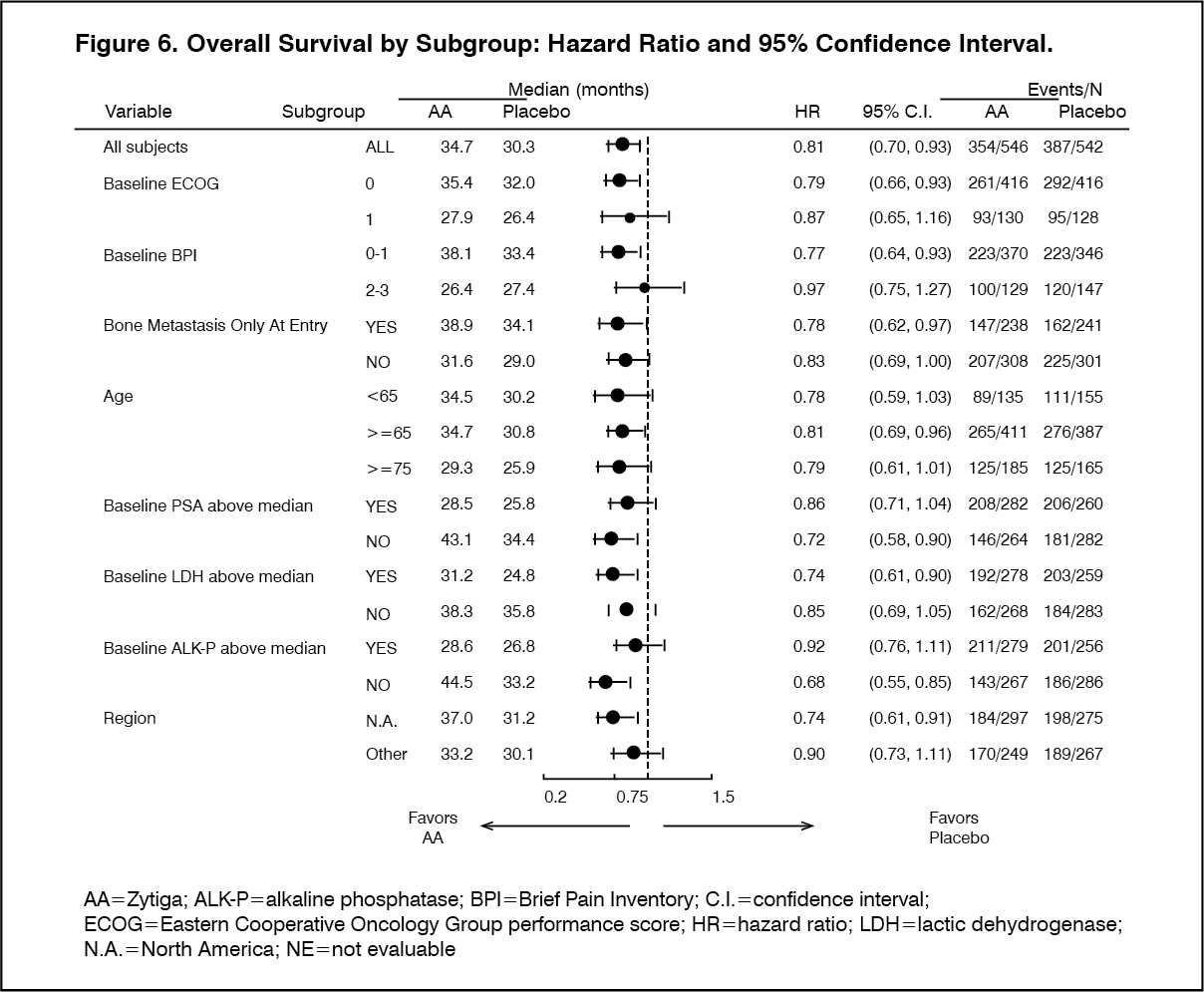
Click on icon to see table/diagram/imageSubgroup analyses consistently favor treatment with ZYTIGA (see Figure 6).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn addition to the observed improvements in overall survival and rPFS, benefit was demonstrated for ZYTIGA vs. placebo treatment in all prospectively defined secondary endpoint measures as follows: Time to PSA progression based on PCWG2 criteria: The median time to PSA progression was 11.1 months for patients receiving ZYTIGA and 5.6 months for patients receiving placebo (HR = 0.488; 95% CI: [0.420, 0.568], p < 0.0001). The time to PSA progression was approximately doubled with ZYTIGA treatment (HR = 0.488). The proportion of subjects with a confirmed PSA response was greater in the ZYTIGA group than in the placebo group (62% versus 24%; p < 0.0001).
Time to opiate use for cancer pain: The median time to opiate use for prostate cancer pain at the time of final analysis was 33.4 months for patients receiving ZYTIGA and was 23.4 months for patients receiving placebo (HR = 0.721; 95% CI: [0.614, 0.846], p ≤ 0.0001).
Time to initiation of cytotoxic chemotherapy: The median time to initiation of cytotoxic chemotherapy was 25.2 months for patients receiving ZYTIGA and 16.8 months for patients receiving placebo (HR = 0.580; 95% CI: [0.487, 0.691], p < 0.0001).
Time to deterioration in ECOG performance score by ≥ 1 point: The median time to deterioration in ECOG performance score by ≥ 1 point was 12.3 months for patients receiving ZYTIGA and 10.9 months for patients receiving placebo (HR = 0.821; 95% CI: [0.714, 0.943], p = 0.0053).
The following study endpoints demonstrated a statistically significant advantage in favor of ZYTIGA treatment: Objective response: Objective response was defined as the proportion of subjects with measurable disease achieving a complete or partial response according to RECIST criteria (baseline lymph node size was required to be ≥ 2 cm to be considered a target lesion). The proportion of subjects with measurable disease at baseline who had an objective response was 36% in the ZYTIGA group and 16% in the placebo group (p < 0.0001).
Pain: Treatment with ZYTIGA significantly reduced the risk of average pain intensity progression by 18% compared with placebo (p=0.0490). The median time to progression was 26.7 months in the ZYTIGA group and 18.4 months in the placebo group.
Time to degradation in the FACT-P (Total Score): Treatment with ZYTIGA decreased the risk of FACT-P (Total Score) degradation by 22% compared with placebo (p = 0.0028). The median time to degradation in FACT-P (Total Score) was 12.7 months in the ZYTIGA group and 8.3 months in the placebo group.
Study 301 (patients who had received prior chemotherapy): Eleven percent of patients enrolled in Study 301 had an ECOG performance score of 2; 70% had radiographic evidence of disease progression with or without PSA progression; 70% had received one prior cytotoxic chemotherapy and 30% received two. Liver metastasis was present in 11% of patients treated with ZYTIGA.
It was recommended that patients be maintained on their study drugs until there was PSA progression (confirmed 25% increase over the patient's baseline/nadir) together with protocol-defined radiographic progression and symptomatic or clinical progression. The primary efficacy endpoint was overall survival.
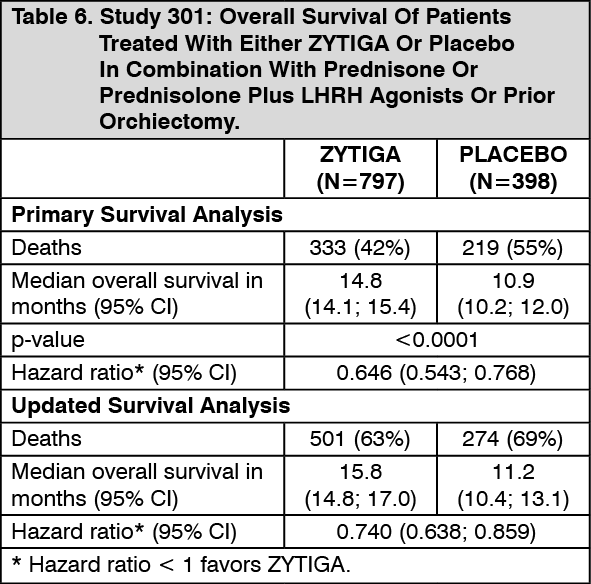
In a planned analysis conducted after 552 deaths were observed, 42% (333 of 797) of patients treated with ZYTIGA, compared with 55% (219 of 398) of patients treated with placebo, had died. A statistically significant improvement in median overall survival was seen in patients treated with ZYTIGA (see Table 6 and Figure 7). An updated survival analysis was conducted when 775 deaths (97% of the planned number of deaths for the final analysis) were observed. Results from this updated survival analysis were consistent with those in the primary survival analysis (see Table 2). (See Table 6.)
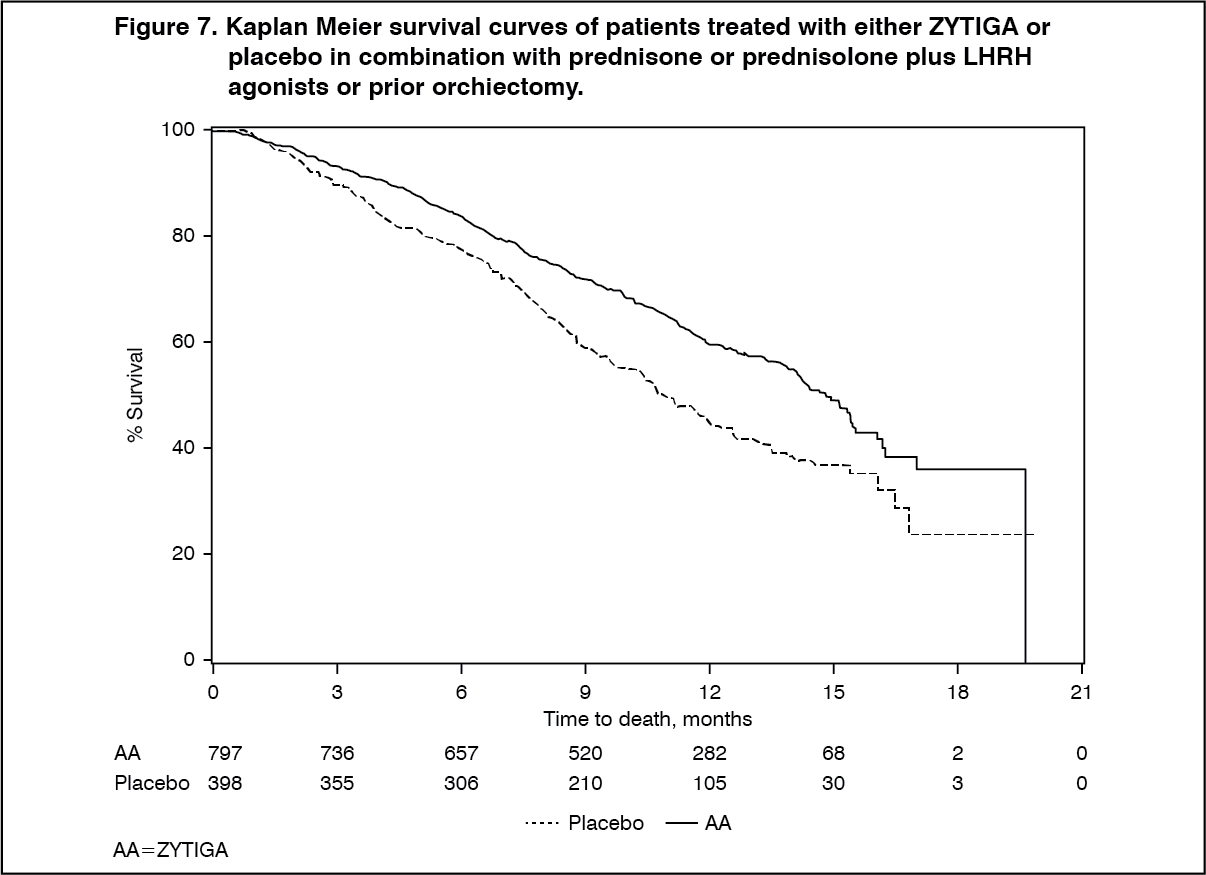
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAt all evaluation time points after the initial few months of treatment, a higher proportion of patients treated with ZYTIGA remained alive, compared with the proportion of patients treated with placebo (see Figure 7).
 Click on icon to see table/diagram/image
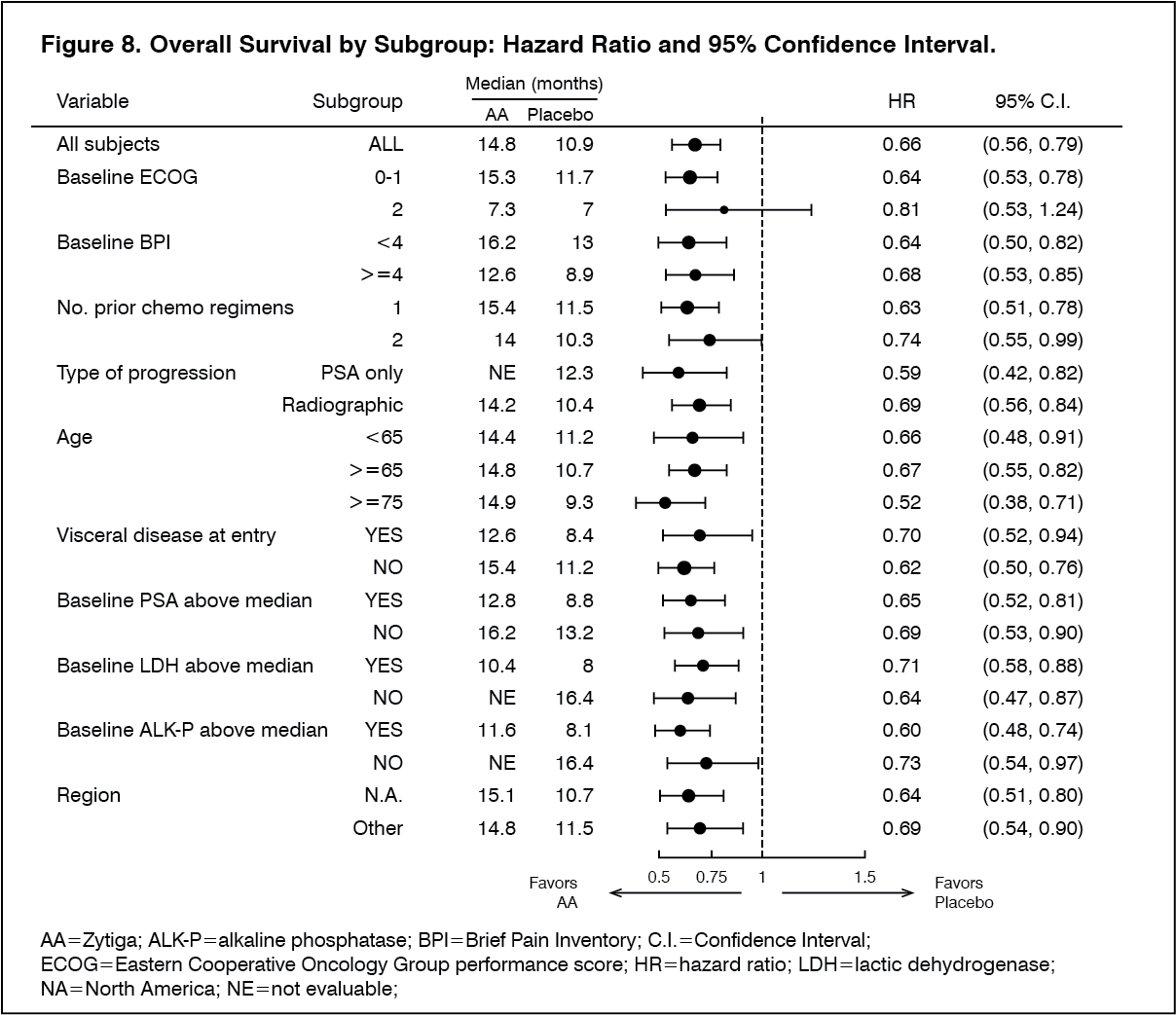
Click on icon to see table/diagram/imageSubgroup survival analyses showed a consistent survival benefit for treatment with ZYTIGA (see Figure 8).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn addition to the observed improvement in overall survival, all secondary study endpoints favored ZYTIGA and were statistically significant after adjusting for multiple testing as follows: Patients receiving ZYTIGA demonstrated a significantly higher total PSA response rate (defined as a ≥ 50% reduction from baseline), compared with patients receiving placebo, 38% versus 10%, p < 0.0001.
The median time to PSA progression was 10.2 months for patients treated with ZYTIGA and 6.6 months for patients treated with placebo (HR = 0.580; 95% CI: [0.462; 0.728], p < 0.0001).
The median radiographic progression-free survival was 5.6 months for patients treated with ZYTIGA and 3.6 months for patients who received placebo (HR = 0.673; 95% CI: [0.585; 0.776], p < 0.0001).
Pain: The proportion of patients with pain palliation was statistically significantly higher in the ZYTIGA group than in the placebo group (44% versus 27%, p = 0.0002). A responder for pain palliation was defined as a patient who experienced at least a 30% reduction from baseline in the BPI-SF worst pain intensity score over the last 24 hours without any increase in analgesic usage score observed at two consecutive evaluations four weeks apart. Only patients with a baseline pain score of ≥ 4 and at least one post-baseline pain score were analyzed (n = 512) for pain palliation.
A lower proportion of patients treated with ZYTIGA had pain progression compared to patients taking placebo at 6 (22% versus 28%), 12 (30% versus 38%) and 18 months (35% versus 46%). Pain progression was defined as an increase from baseline of ≥ 30% in the BPI-SF worst pain intensity score over the previous 24 hours without a decrease in analgesic usage score observed at two consecutive visits, or an increase of ≥ 30% in analgesic usage score observed at two consecutive visits. The time to pain progression at the 25th percentile was 7.4 months in the ZYTIGA group, versus 4.7 months in the placebo group.
Skeletal-Related events: A lower proportion of patients in the ZYTIGA group had skeletal-related events compared with the placebo group at 6 months (18% vs. 28%), 12 months (30% vs. 40%), and 18 months (35% vs. 40%). The time to first skeletal-related event at the 25th percentile in the ZYTIGA group was twice that of the control group at 9.9 months vs. 4.9 months. A skeletal-related event was defined as a pathological fracture, spinal cord compression, palliative radiation to bone, or surgery to bone.
Pharmacokinetics: General introduction: Following administration of abiraterone acetate, the pharmacokinetics of abiraterone and abiraterone acetate have been studied in healthy subjects, patients with metastatic advanced prostate cancer and subjects without cancer with hepatic or renal impairment. Abiraterone acetate is rapidly converted in vivo to abiraterone, an androgen biosynthesis inhibitor (see Pharmacology: Pharmacodynamics: Mechanism of Action under Actions).
Absorption: Following oral administration of abiraterone acetate in the fasting state, the time to reach maximum plasma abiraterone concentration is approximately 2 hours.
Administration of abiraterone acetate with food, compared with administration in a fasted state, results in up to a 17-fold increase in mean systemic exposure of abiraterone, depending on the fat content of the meal. Given the normal variation in the content and composition of meals, taking ZYTIGA with meals has the potential to result in highly variable exposures. Therefore, ZYTIGA must not be taken with food. ZYTIGA tablets must be taken as a single dose once daily on an empty stomach. ZYTIGA should be taken at least two hours after eating and no food should be eaten for at least 1 hour after taking ZYTIGA. The tablets should be swallowed whole with water (see Dosage & Administration).
Distribution and protein binding: The plasma protein binding of 14C-abiraterone in human plasma is 99.8%. The apparent volume of distribution is approximately 5630 L, suggesting that abiraterone extensively distributes to peripheral tissues.
Metabolism: Following oral administration of 14C-abiraterone acetate as capsules, abiraterone acetate is hydrolyzed to abiraterone, which then undergoes metabolism including sulphation, hydroxylation and oxidation primarily in the liver. The majority of circulating radioactivity (approximately 92%) is found in the form of metabolites of abiraterone. Of 15 detectable metabolites, 2 main metabolites, abiraterone sulphate and N-oxide abiraterone sulphate, each represents approximately 43% of total radioactivity.
Elimination: The mean half-life of abiraterone in plasma is approximately 15 hours based on data from healthy subjects. Following oral administration of 14C-abiraterone acetate, approximately 88% of the radioactive dose is recovered in faeces and approximately 5% in urine. The major compounds present in feces are unchanged abiraterone acetate and abiraterone (approximately 55% and 22% of the administered dose, respectively).
Special populations: Renal impairment: The pharmacokinetics of abiraterone was compared in patients with end-stage renal disease on a stable hemodialysis schedule, versus matched control subjects with normal renal function. Systemic exposure to abiraterone after a single oral 1000 mg dose did not increase in patients with end-stage renal disease on dialysis.
Administration of ZYTIGA in patients with renal impairment including severe renal impairment does not require dose reduction (see Dosage & Administration).
Hepatic impairment: The pharmacokinetics of abiraterone acetate was examined in subjects with pre-existing mild or moderate hepatic impairment (Child-Pugh Class A and B, respectively) and in healthy control subjects. Systemic exposure to abiraterone after a single oral 1,000 mg dose increased by approximately 11% and 260% in subjects with mild and moderate pre-existing hepatic impairment, respectively. The mean half-life of abiraterone is prolonged to approximately 18 hours in subjects with mild hepatic impairment and to approximately 19 hours in subjects with moderate hepatic impairment. No dose adjustment is necessary for patients with pre-existing mild hepatic impairment. There are no data on the clinical safety and efficacy of multiple doses of abiraterone acetate when administered to patients with moderate or severe hepatic impairment (Child Pugh Class B or C). No dosage adjustment can be predicted. ZYTIGA should be used with caution in patients with moderate hepatic impairment, only if the benefit clearly outweighs the possible risk (see Dosage & Administration and Hepatotoxicity and Hepatic impairment under Precautions).
Effects on the QT interval: In a cardiovascular safety study in patients with metastatic advanced prostate cancer there were no significant effects of abiraterone acetate on the cardiac QT/QTc interval.
Toxicology: Non-Clinical Information: Carcinogenicity and mutagenicity: Abiraterone acetate was not carcinogenic in a 6-month study in the transgenic (Tg.rasH2) mouse. In a 24-month carcinogenicity study in the rat, abiraterone acetate increased the incidence of interstitial cell neoplasms in the testes. This finding is considered related to the pharmacological action of abiraterone and rat specific. Abiraterone acetate was not carcinogenic in female rats.
Abiraterone acetate and abiraterone were devoid of genotoxic potential in the standard panel of genotoxicity tests, including an in vitro bacterial reverse mutation assay (the Ames test), an in vitro mammalian chromosome aberration test (using human lymphocytes) and an in vivo rat micronucleus assay.
Reproductive toxicology: In fertility studies in both male and female rats, abiraterone acetate reduced fertility, which was completely reversible in 4 to 16 weeks after abiraterone acetate was stopped.
In a developmental toxicity study in the rat, abiraterone acetate affected pregnancy including reduced fetal weight and survival. Effects on the external genitalia were observed though abiraterone acetate was not teratogenic.
In these fertility and developmental toxicity studies performed in the rat, all effects were related to the pharmacological activity of abiraterone.
ZYTIGA is contraindicated in pregnancy (see Contraindications and Use in Pregnancy & Lactation).
Animal toxicology: In all animal toxicity studies, circulating testosterone levels were significantly reduced. As a result, reduction in organ weights and morphological and/or histopathological changes in the reproductive organs, and the adrenal, pituitary and mammary glands were observed. All changes showed complete or partial reversibility. The changes in the reproductive organs and androgen-sensitive organs are consistent with the pharmacology of abiraterone. All treatment-related hormonal changes reversed or were shown to be resolving after a 4-week recovery period.