Therapeutic Class: VYTORIN (ezetimibe/simvastatin) is a lipid-lowering product that selectively inhibits the intestinal absorption of cholesterol and related plant sterols and inhibits the endogenous synthesis of cholesterol.
Pharmacology: Pharmacodynamics: Mechanism of Action: VYTORIN: Plasma cholesterol is derived from intestinal absorption and endogenous synthesis. VYTORIN contains ezetimibe and simvastatin, two lipid-lowering compounds with complementary mechanisms of action. VYTORIN reduces elevated total-C, LDL-C, Apo B, TG, and non-HDL-C, and increases HDL-C through dual inhibition of cholesterol absorption and synthesis.
Ezetimibe: Ezetimibe inhibits the intestinal absorption of cholesterol. Ezetimibe is orally active and has a mechanism of action that differs from other classes of cholesterol-reducing compounds (e.g., statins, bile acid sequestrants [resins], fibric acid derivatives, and plant stanols). The molecular target of ezetimibe is the sterol transporter, Niemann-Pick C1-Like 1 (NPC1L1), which is responsible for the intestinal uptake of cholesterol and phytosterols.
Ezetimibe localizes at the brush border of the small intestine and inhibits the absorption of cholesterol, leading to a decrease in the delivery of intestinal cholesterol to the liver; statins reduce cholesterol synthesis in the liver and together these distinct mechanisms provide complementary cholesterol reduction.
In a 2-week clinical study in 18 hypercholesterolemic patients, ezetimibe inhibited intestinal cholesterol absorption by 54%, compared with placebo.
A series of preclinical studies was performed to determine the selectivity of ezetimibe for inhibiting cholesterol absorption. Ezetimibe inhibited the absorption of [
14C]-cholesterol with no effect on the absorption of triglycerides, fatty acids, bile acids, progesterone, ethinyl estradiol, or the fat-soluble vitamins A and D.
Simvastatin: After oral ingestion, simvastatin, which is an inactive lactone, is hydrolyzed in the liver to the corresponding active β-hydroxyacid form which has a potent activity in inhibiting HMG-CoA reductase (3 hydroxy-3 methylglutaryl CoA reductase). This enzyme catalyses the conversion of HMG-CoA to mevalonate, an early and rate-limiting step in the biosynthesis of cholesterol.
Simvastatin has been shown to reduce both normal and elevated LDL-C concentrations. LDL is formed from very-low-density protein (VLDL) and is catabolized predominantly by the high affinity LDL receptor. The mechanism of the LDL-lowering effect of simvastatin may involve both reduction of VLDL-cholesterol (VLDL-C) concentration and induction of the LDL receptor, leading to reduced production and increased catabolism of LDL-C. Apolipoprotein B also falls substantially during treatment with simvastatin. In addition, simvastatin moderately increases HDL-C and reduces plasma TG. As a result of these changes, the ratios of total- to HDL-C and LDL- to HDL-C are reduced.
Clinical Studies: In controlled clinical studies, VYTORIN significantly reduced total cholesterol (total-C), low-density lipoprotein cholesterol (LDL-C), apolipoprotein B (Apo B), triglycerides (TG), and non-high-density lipoprotein cholesterol (non-HDL-C), and increased high-density lipoprotein cholesterol (HDL-C) in patients with hypercholesterolemia.
VYTORIN: Prevention of Cardiovascular Disease: The IMProved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT) was a multicenter, randomized, double-blind, active-control study of 18,144 patients enrolled within 10 days of hospitalization for acute coronary syndrome (ACS; either acute myocardial infarction [MI] or unstable angina [UA]). Patients had an LDL-C ≤125 mg/dL (≤3.2 mmol/L) at the time of presentation with ACS if they had not been taking lipid-lowering therapy, or ≤100 mg/dL (≤2.6 mmol/L) if they had been receiving lipid-lowering therapy. All patients were randomized in a 1:1 ratio to receive either VYTORIN 10/40 mg (n=9067) or simvastatin 40 mg (n=9077) and followed for a median of 6.0 years.
Patients had a mean age of 63.6 years; 76% were male, 84% were Caucasian, and 27% were diabetic. The average LDL-C value at the time of study qualifying event was 80 mg/dL (2.1 mmol/L) for those on lipid-lowering therapy (n=6390) and 101 mg/dL (2.6 mmol/L) for those not on previous lipid-lowering therapy (n=11594). Prior to the hospitalization for the qualifying ACS event, 34% of the patients were on statin therapy. At one year, the average LDL-C for patients continuing on therapy was 53.2 mg/dL (1.4 mmol/L) for the VYTORIN group and 69.9 mg/dL (1.8 mmol/L) for the simvastatin monotherapy group. Lipid values were generally obtained for patients who remained on study therapy.
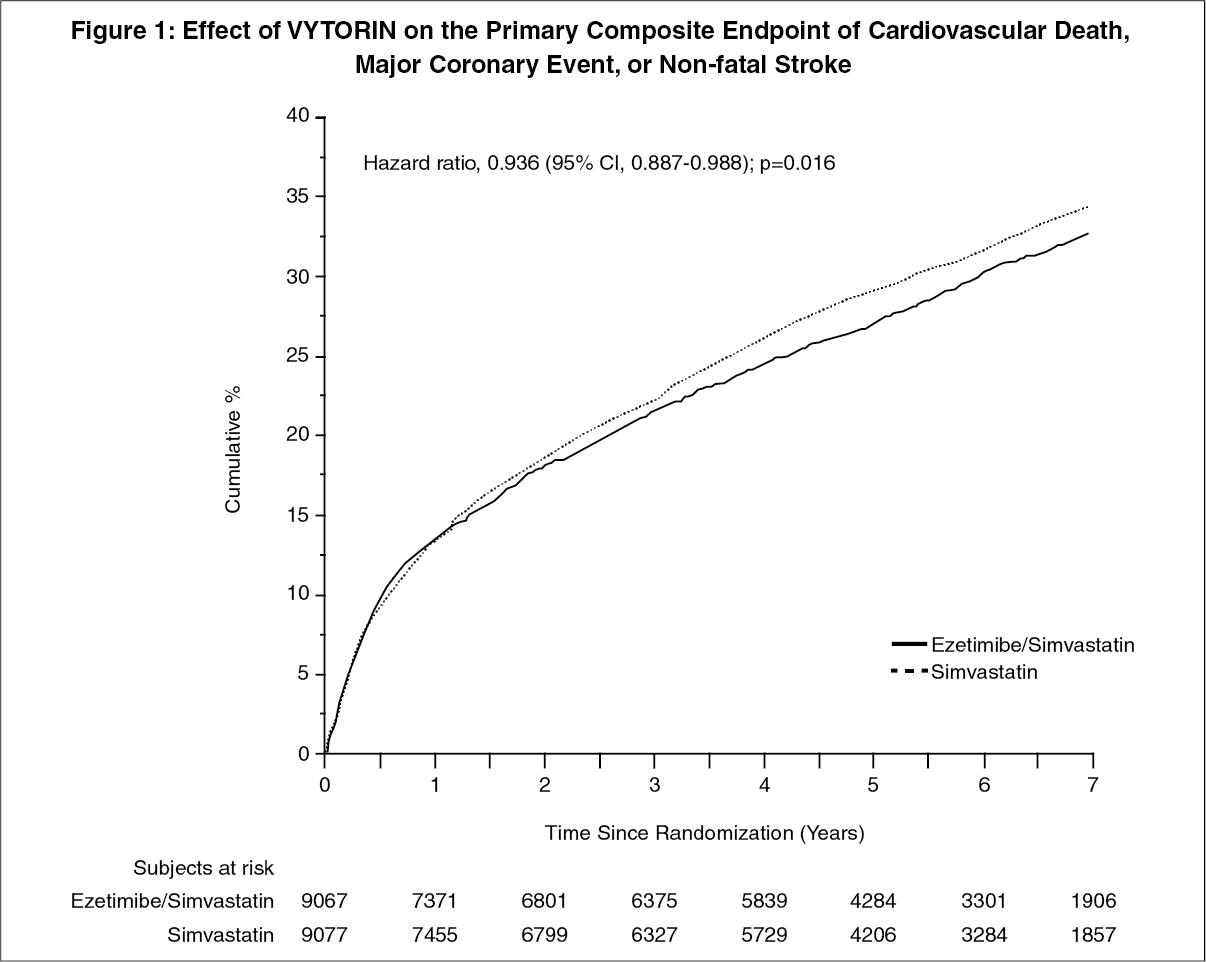
The primary endpoint was a composite consisting of cardiovascular death, major coronary events (MCE; defined as non-fatal myocardial infarction, documented unstable angina that required hospitalization, or any coronary revascularization procedure occurring at least 30 days after randomized treatment assignment) and non-fatal stroke. The study demonstrated that treatment with VYTORIN provided incremental benefit in reducing the primary composite endpoint of cardiovascular death, MCE, and non-fatal stroke compared with simvastatin alone (relative risk reduction of 6.4%, p=0.016). The primary endpoint occurred in 2572 of 9067 patients (7-year Kaplan-Meier [KM] rate 32.72%) in the VYTORIN group and 2742 of 9077 patients (7-year KM rate 34.67%) in the simvastatin alone group. (See Figure 1 and Table 1.)
The treatment effect of VYTORIN was generally consistent with the overall results across many subgroups, including sex, age, race, medical history of diabetes mellitus, baseline lipid levels, prior statin therapy, prior stroke, and hypertension (see Figure 2). (See Figures 1 and 2, Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Primary Hypercholesterolemia: VYTORIN: Five multicenter, double-blind studies conducted with VYTORIN in patients with primary hypercholesterolemia are reported: two were comparisons with simvastatin, two were comparisons with atorvastatin, and one was a comparison with rosuvastatin.
In a multicenter, double-blind, placebo-controlled, 12-week trial, 887 hypercholesterolemic patients were randomized to one of ten treatment groups: placebo, ezetimibe (10 mg), simvastatin (10 mg, 20 mg, 40 mg, or 80 mg), or coadministered ezetimibe and simvastatin equivalent to VYTORIN (10/10, 10/20, 10/40, and 10/80). When patients receiving VYTORIN were compared to those receiving all doses of simvastatin, VYTORIN significantly lowered total-C, LDL-C, Apo B, TG, non-HDL-C, and C-reactive protein. The effects of VYTORIN on HDL-C were similar to the effects seen with simvastatin. Further analysis showed VYTORIN significantly increased HDL-C compared with placebo. (See Table 2.)
Click on icon to see table/diagram/image
In a similarly designed study, results for all lipid parameters were generally consistent. In a pooled analysis of these two studies, the lipid response to VYTORIN was similar in patients with TG levels greater than or less than 200 mg/dL.
In a multicenter, double-blind, controlled, 23-week study, 710 patients with known CHD or CHD risk equivalents, as defined by the NCEP ATP III guidelines, and an LDL-C ≥130 mg/dL were randomized to one of four treatment groups: coadministered ezetimibe and simvastatin equivalent to VYTORIN (10/10, 10/20, and 10/40), or simvastatin 20 mg. Patients not reaching an LDL-C <100 mg/dL had their simvastatin dose titrated at 6-week intervals to a maximal dose of 80 mg. At Week 5, the LDL-C reductions with VYTORIN 10/10, 10/20, or 10/40 were significantly larger than with simvastatin 20 mg. In addition, at Week 5, significantly more patients receiving VYTORIN 10/10, 10/20, or 10/40 attained LDL-C target compared to those receiving simvastatin 20 mg (see Table 3). Week 5 results for LDL-C reduction and percentage attaining LDL-C target were consistent with the end of study results (Week 23). (See Table 3.)
Click on icon to see table/diagram/image
In a multicenter, double-blind, 6-week study, 1902 patients with primary hypercholesterolemia, who had not met their NCEP ATP III target LDL-C goal, were randomized to one of eight treatment groups: VYTORIN (10/10, 10/20, 10/40, or 10/80) or atorvastatin (10 mg, 20 mg, 40 mg, or 80 mg). When patients receiving all doses of VYTORIN were compared to those receiving all doses of atorvastatin, VYTORIN lowered total-C, LDL-C, Apo B, and non-HDL-C, and increased HDL-C significantly more than atorvastatin. The effects of VYTORIN on TG were similar to the effects seen with atorvastatin. (See Table 4.)
Click on icon to see table/diagram/image
In a multicenter, double-blind, 24-week, forced titration study, 788 patients with primary hypercholesterolemia, who had not met their NCEP ATP III target LDL-C goal, were randomized to receive coadministered ezetimibe and simvastatin equivalent to VYTORIN (10/10 and 10/20) or atorvastatin 10 mg. For all three treatment groups, the dose of the statin was titrated at 6-week intervals to 80 mg. At each pre-specified dose comparison, VYTORIN lowered LDL-C to a greater degree than atorvastatin (see Table 5).
Click on icon to see table/diagram/image
In a multicenter, double-blind, 6-week study, 2959 patients with primary hypercholesterolemia, who had not met their NCEP ATP III target LDL-C goal, were randomized to one of six treatment groups: VYTORIN (10/20, 10/40, or 10/80) or rosuvastatin (10 mg, 20 mg, or 40 mg). When patients receiving all doses of VYTORIN were compared to those receiving all doses of rosuvastatin, VYTORIN lowered total-C, LDL-C, Apo B, TG, and non-HDL-C significantly more than rosuvastatin. The effects of VYTORIN on HDL-C were similar to the effects seen with rosuvastatin (see Table 6).
Click on icon to see table/diagram/image
In a double-blind, placebo-controlled, 8-week study, 240 patients with hypercholesterolemia already receiving simvastatin monotherapy and not at National Cholesterol Education Program (NCEP) LDL-C goal (2.6 to 4.1 mmol/L [100 to 160 mg/dL], depending on baseline characteristics) were randomized to receive either ezetimibe 10 mg or placebo in addition to their on-going simvastatin therapy. Among simvastatin-treated patients not at LDL-C goal at baseline (~80%), significantly more patients randomized to ezetimibe coadministered with simvastatin achieved their LDL-C goal at study endpoint compared to patients randomized to placebo coadministered with simvastatin, 76% and 21.5%, respectively. The corresponding LDL-C reductions for ezetimibe or placebo coadministered with simvastatin were also significantly different (27% or 3%, respectively). In addition, ezetimibe coadministered with simvastatin significantly decreased total-C, Apo B, and TG compared with placebo coadministered with simvastatin.
In a multicenter, double-blind, 24-week trial, 214 patients with type 2 diabetes mellitus treated with thiazolidinediones (rosiglitazone or pioglitazone) for a minimum of 3 months and simvastatin 20 mg for a minimum of 6 weeks with a mean LDL-C of 93 mg/dL, were randomized to receive either simvastatin 40 mg or the coadministered active ingredients equivalent to VYTORIN 10/20.
VYTORIN 10/20 was significantly more effective than doubling the dose of simvastatin to 40 mg in further reducing LDL-C (-21% and 0%, respectively), total-C (-14% and -1%, respectively), Apo B (-14% and -2%, respectively), and non-HDL-C (-20% and -2%, respectively) beyond the reductions observed with simvastatin 20 mg. Results for HDL-C and TG between the two treatment groups were not significantly different. Results were not affected by type of thiazolidinedione treatment.
Ezetimibe: In two, multicenter, double-blind, placebo-controlled, 12-week studies in 1719 patients with primary hypercholesterolemia, ezetimibe significantly lowered total-C (13%), LDL-C (19%), Apo B (14%), and TG (8%) and increased HDL-C (3%) compared to placebo. Reduction in LDL-C was consistent across age, sex, race, and baseline LDL-C. In addition, ezetimibe had no effect on the plasma concentrations of the fat-soluble vitamins A, D, and E, had no effect on prothrombin time, and did not impair adrenocortical steroid hormone production.
Simvastatin: VYTORIN contains simvastatin. In two large, placebo-controlled clinical trials, the Scandinavian Simvastatin Survival Study (N=4,444 patients) and the Heart Protection Study (N=20,536 patients), the effects of treatment with simvastatin were assessed in patients at high risk of coronary events because of existing coronary heart disease, diabetes, peripheral vessel disease, history of stroke or other cerebrovascular disease. Simvastatin was proven to reduce: the risk of total mortality by reducing CHD deaths, the risk of non-fatal myocardial infarction and stroke, and the need for coronary and non-coronary revascularization procedures.
Homozygous Familial Hypercholesterolemia (HoFH): A double-blind, randomized, 12-week study was performed in patients with a clinical and/or genotypic diagnosis of HoFH. Data were analyzed from a subgroup of patients (n=14) receiving simvastatin 40 mg at baseline. Increasing the dose of simvastatin from 40 to 80 mg (n=5) produced a reduction of LDL-C of 13% from baseline on simvastatin 40 mg. Coadministered ezetimibe and simvastatin equivalent to VYTORIN (10/40 and 10/80 pooled, n=9), produced a reduction of LDL-C of 23% from baseline on simvastatin 40 mg. In those patients coadministered ezetimibe and simvastatin equivalent to VYTORIN (10/80, n=5), a reduction of LDL-C of 29% from baseline on simvastatin 40 mg was produced.
Pharmacokinetics: Absorption: Ezetimibe: After oral administration, ezetimibe is rapidly absorbed and extensively conjugated to a pharmacologically active phenolic glucuronide (ezetimibe-glucuronide). Mean maximum plasma concentrations (C
max) occur within 1 to 2 hours for ezetimibe-glucuronide and 4 to 12 hours for ezetimibe. The absolute bioavailability of ezetimibe cannot be determined as the compound is virtually insoluble in aqueous media suitable for injection.
Concomitant food administration (high fat or non-fat meals) had no effect on the oral bioavailability of ezetimibe when administered as ezetimibe 10-mg tablets.
Simvastatin: The availability of the β-hydroxyacid to the systemic circulation following an oral dose of simvastatin was found to be less than 5% of the dose, consistent with extensive hepatic first-pass extraction. The major metabolites of simvastatin present in human plasma are the β-hydroxyacid and four additional active metabolites.
Relative to the fasting state, the plasma profiles of both active and total inhibitors were not affected when simvastatin was administered immediately before a test meal.
Distribution: Ezetimibe: Ezetimibe and ezetimibe-glucuronide are bound 99.7% and 88 to 92% to human plasma proteins, respectively.
Simvastatin: Both simvastatin and the β-hydroxyacid are bound to human plasma proteins (95%).
The pharmacokinetics of single and multiple doses of simvastatin showed that no accumulation of drug occurred after multiple dosing. In all of the above pharmacokinetic studies, the maximum plasma concentration of inhibitors occurred 1.3 to 2.4 hours post-dose.
Metabolism: Ezetimibe: Ezetimibe is metabolized primarily in the small intestine and liver via glucuronide conjugation (a phase II reaction) with subsequent biliary excretion. Minimal oxidative metabolism (a phase I reaction) has been observed in all species evaluated. Ezetimibe and ezetimibe-glucuronide are the major drug-derived compounds detected in plasma, constituting approximately 10 to 20% and 80 to 90% of the total drug in plasma, respectively. Both ezetimibe and ezetimibe-glucuronide are slowly eliminated from plasma with evidence of significant enterohepatic recycling. The half-life for ezetimibe and ezetimibe-glucuronide is approximately 22 hours.
Simvastatin: Simvastatin is an inactive lactone which is readily hydrolyzed
in vivo to the corresponding β-hydroxyacid, a potent inhibitor of HMG-CoA reductase. Hydrolysis takes place mainly in the liver; the rate of hydrolysis in human plasma is very slow.
In man, simvastatin is well absorbed and undergoes extensive hepatic first-pass extraction. The extraction in the liver is dependent on the hepatic blood flow. The liver is its primary site of action, with subsequent excretion of drug equivalents in the bile. Consequently, availability of active drug to the systemic circulation is low.
Following an intravenous injection of the β-hydroxyacid metabolite, its half-life averaged 1.9 hours.
Elimination: Ezetimibe: Following oral administration of
14C-ezetimibe (20 mg) to human subjects, total ezetimibe accounted for approximately 93% of the total radioactivity in plasma. Approximately 78% and 11% of the administered radioactivity were recovered in the feces and urine, respectively, over a 10-day collection period. After 48 hours, there were no detectable levels of radioactivity in the plasma.
Simvastatin: Following an oral dose of radioactive simvastatin to man, 13% of the radioactivity was excreted in the urine and 60% in the feces within 96 hours. The amount recovered in the feces represents absorbed drug equivalents excreted in bile as well as unabsorbed drug. Following an intravenous injection of the β-hydroxyacid metabolite an average of only 0.3% of the IV dose was excreted in urine as inhibitors.
Characteristics in Patients (Special Populations): Pediatric Patients: The absorption and metabolism of ezetimibe are similar between children and adolescents (10 to 18 years) and adults. Based on total ezetimibe, there are no pharmacokinetic differences between adolescents and adults. Pharmacokinetic data in the pediatric population <10 years of age are not available.
Geriatric Patients: Plasma concentrations for total ezetimibe are about 2-fold higher in the elderly (≥65 years) than in the young (18 to 45 years). LDL-C reduction and safety profile are comparable between elderly and young subjects treated with ezetimibe.
Hepatic Insufficiency: After a single 10-mg dose of ezetimibe, the mean area under the curve (AUC) for total ezetimibe was increased approximately 1.7-fold in patients with mild hepatic insufficiency (Child-Pugh score 5 or 6), compared to healthy subjects. In a 14-day, multiple-dose study (10 mg daily) in patients with moderate hepatic insufficiency (Child-Pugh score 7 to 9), the mean AUC for total ezetimibe was increased approximately 4-fold on Day 1 and Day 14 compared to healthy subjects. No dosage adjustment is necessary for patients with mild hepatic insufficiency. Due to the unknown effects of the increased exposure to ezetimibe in patients with moderate or severe (Child-Pugh score >9) hepatic insufficiency, ezetimibe is not recommended in these patients (see Precautions).
Renal Insufficiency: Ezetimibe: After a single 10-mg dose of ezetimibe in patients with severe renal disease (n=8; mean CrCl ≤30 mL/min/1.73 m
2), the mean AUC for total ezetimibe was increased approximately 1.5-fold, compared to healthy subjects (n=9).
An additional patient in this study (post-renal transplant and receiving multiple medications, including cyclosporine) had a 12-fold greater exposure to total ezetimibe.
Simvastatin: In a study of patients with severe renal insufficiency (creatinine clearance <30 mL/min), the plasma concentrations of total inhibitors after a single dose of a related HMG-CoA reductase inhibitor were approximately two-fold higher than those in healthy volunteers.
Gender: Plasma concentrations for total ezetimibe are slightly higher (<20%) in women than in men. LDL-C reduction and safety profile are comparable between men and women treated with ezetimibe.
Race: Based on a meta-analysis of pharmacokinetic studies with ezetimibe, there were no pharmacokinetic differences between Blacks and Caucasians.
Drug Interactions: Diltiazem: In a pharmacokinetic study, concomitant administration of diltiazem caused a 2.7-fold increase in exposure of simvastatin acid, presumably due to inhibition of CYP3A4.
Amlodipine: In a pharmacokinetic study, concomitant administration of amlodipine caused a 1.6-fold increase in exposure of simvastatin acid.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image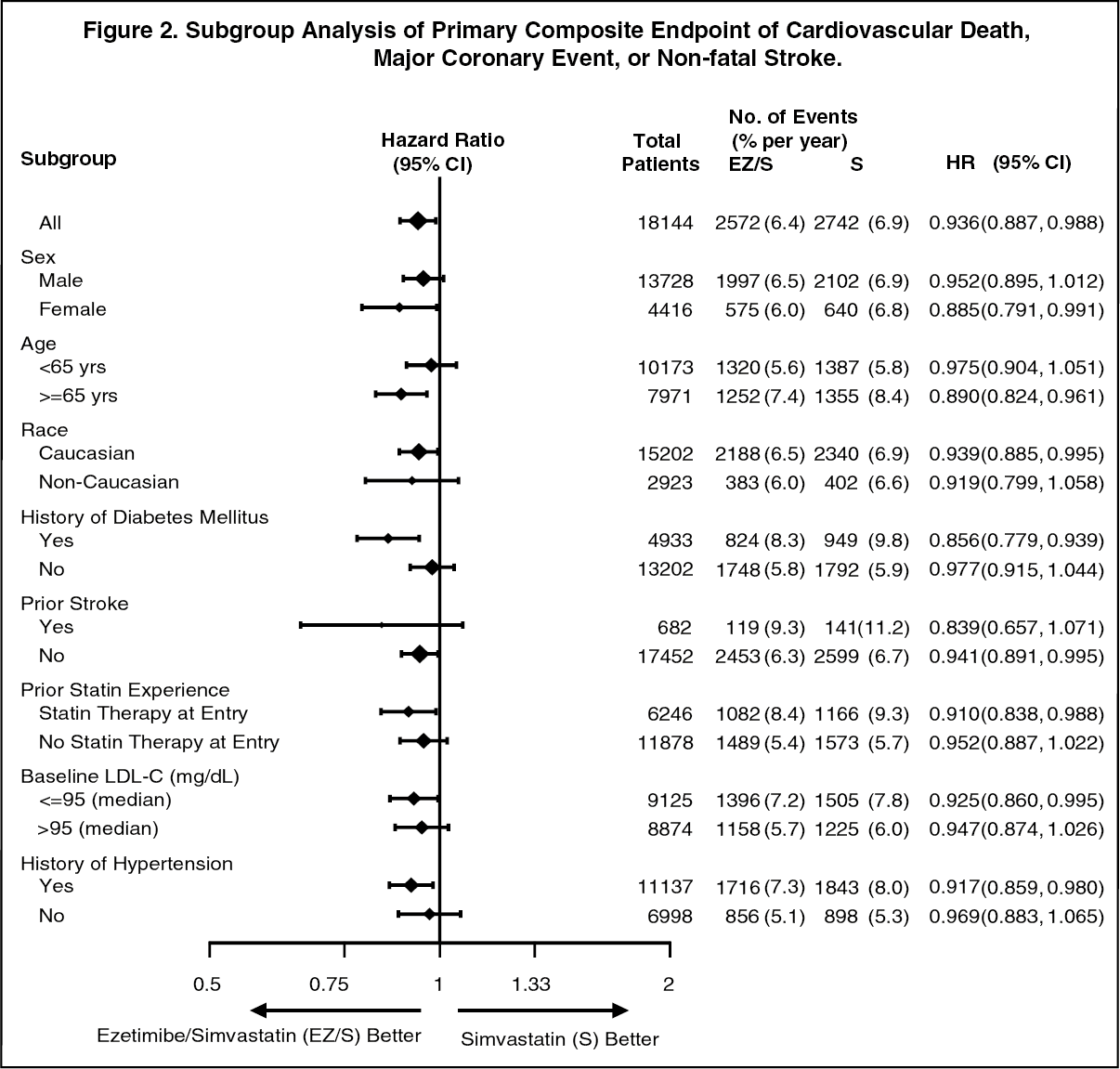 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image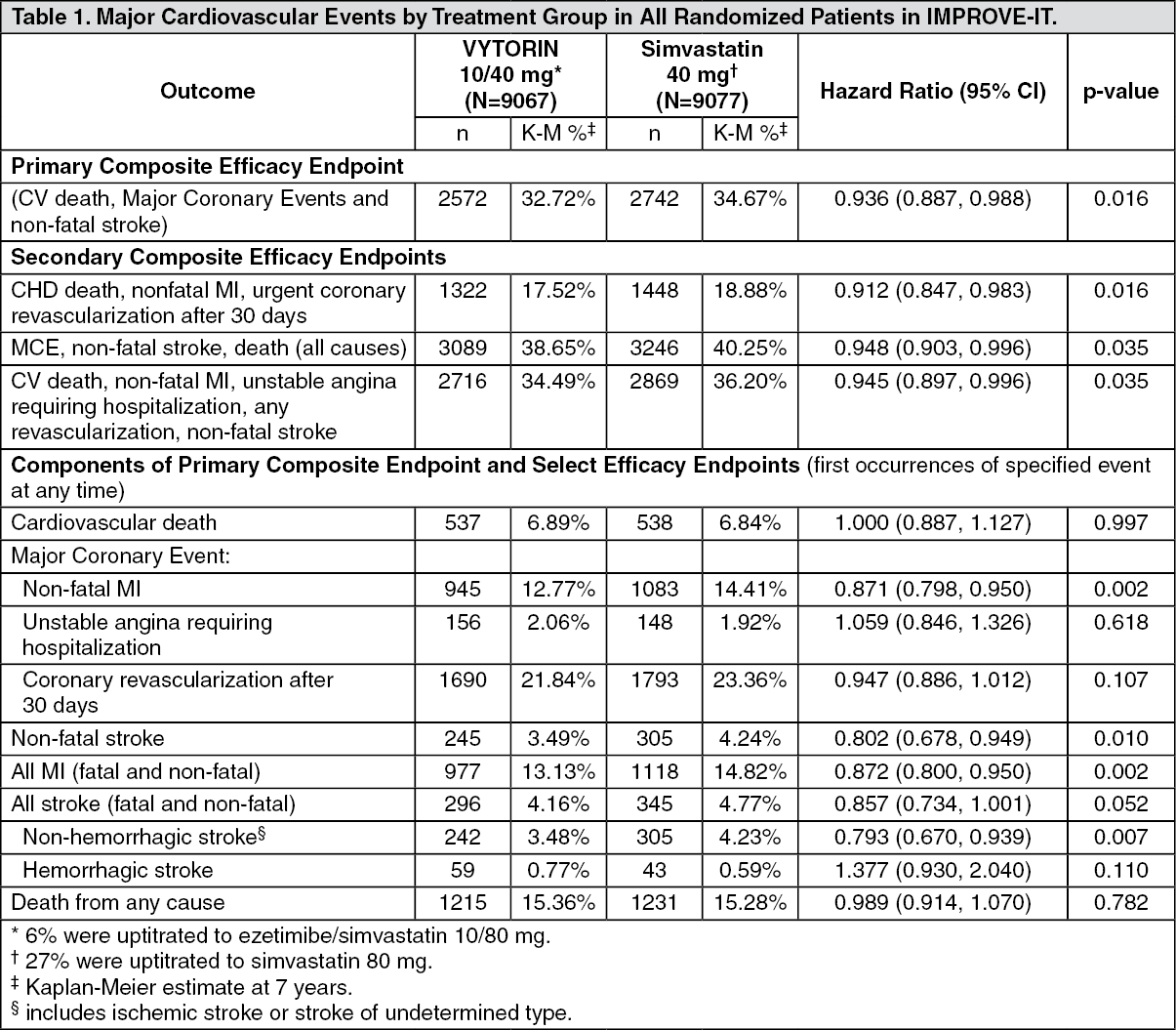 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image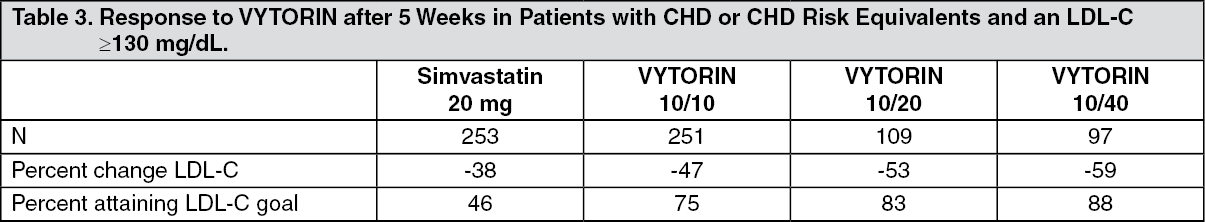 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image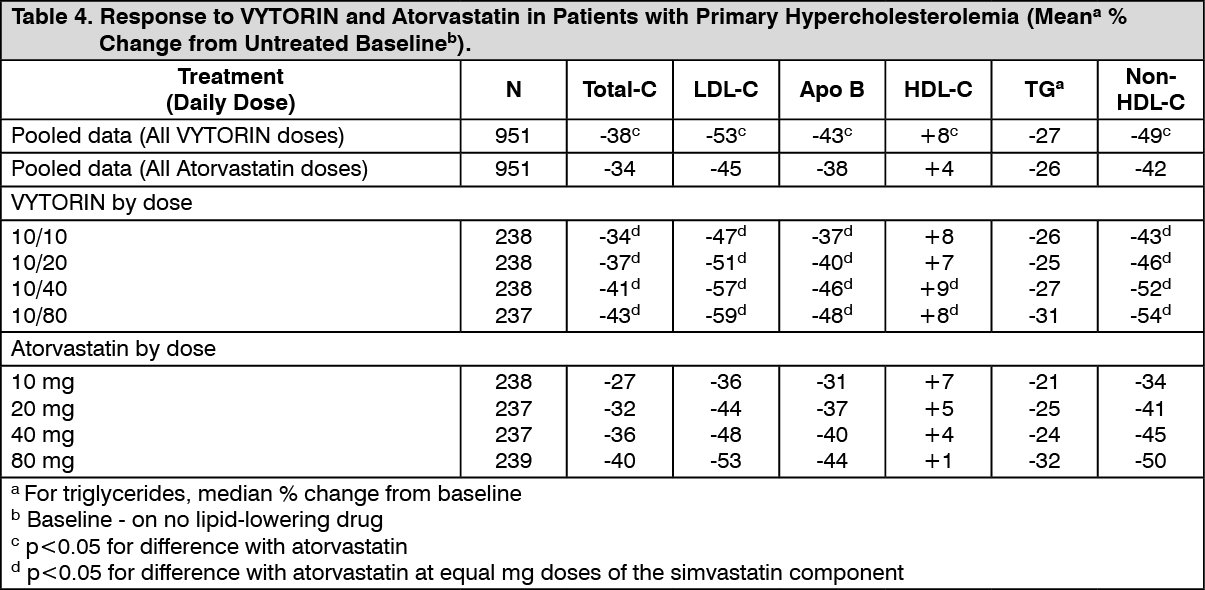 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image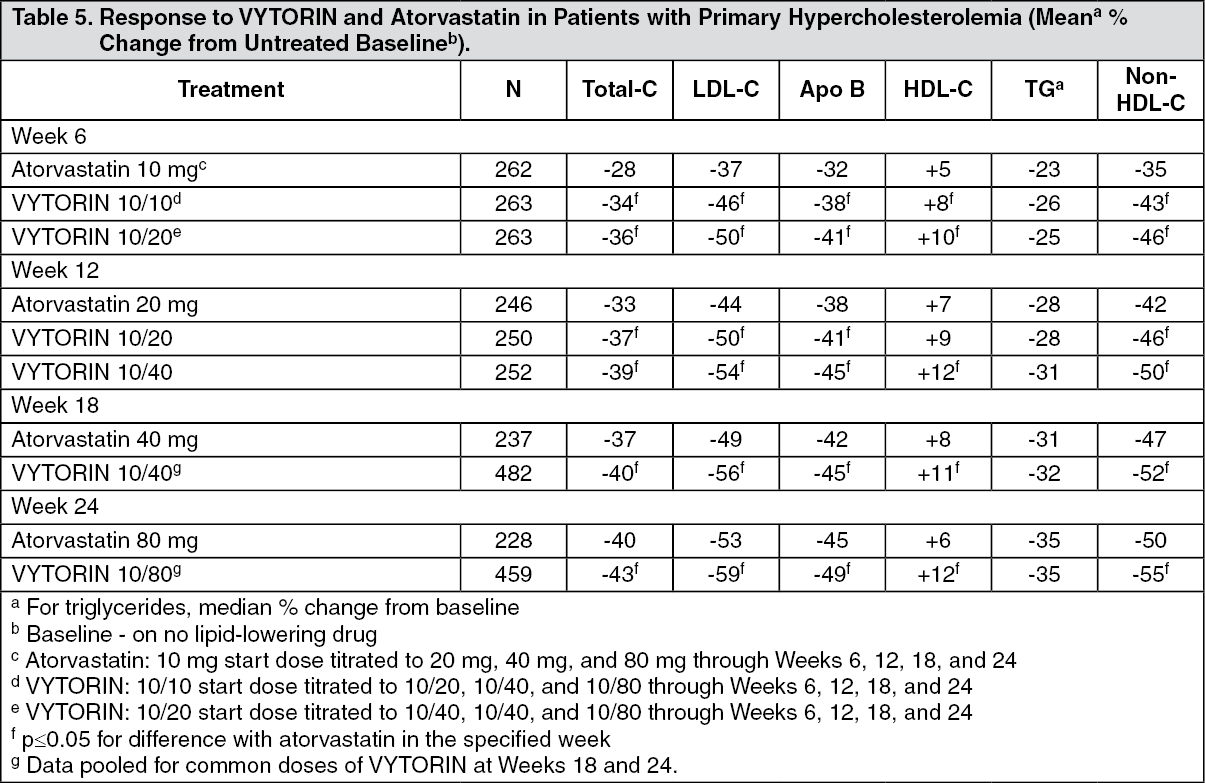 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image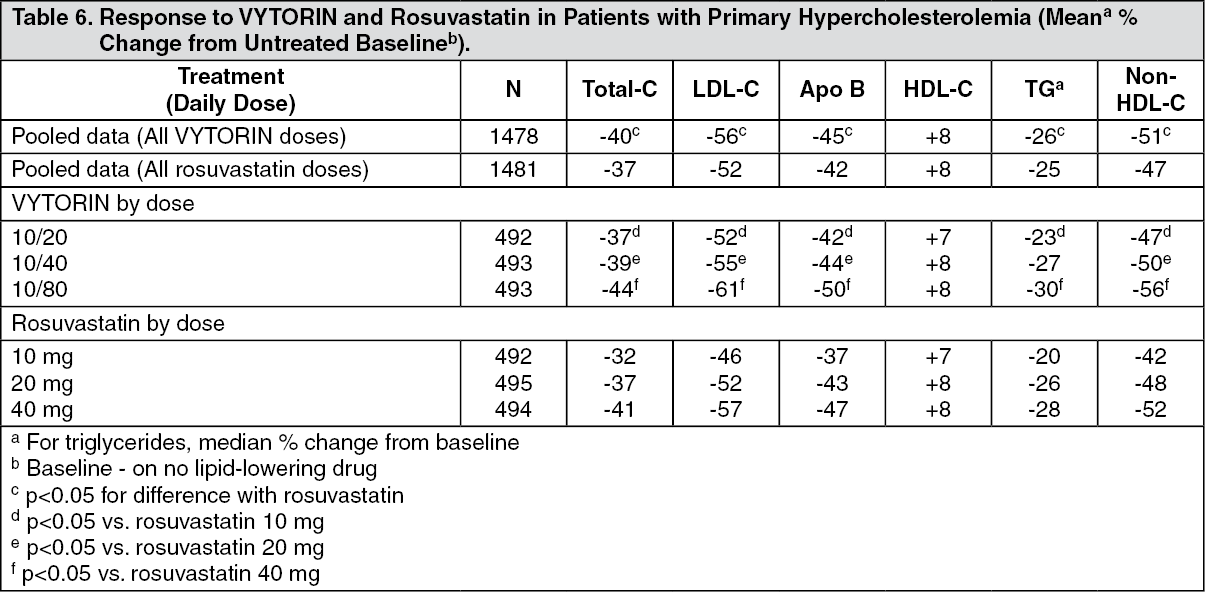 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image



 Sign Out
Sign Out
