


 Sign Out
Sign Out

Pharmacology: Pharmacodynamics: Mechanism of action: Sofosbuvir is a pan-genotypic inhibitor of the HCV NS5B RNA-dependent RNA polymerase, which is essential for viral replication. Sofosbuvir is a nucleotide prodrug that undergoes intracellular metabolism to form the pharmacologically active uridine analog triphosphate (GS-461203), which can be incorporated into HCV RNA by the NS5B polymerase and acts as a chain terminator. In a biochemical assay, GS-461203 inhibited the polymerase activity of the recombinant NS5B from HCV genotype 1b, 2a, 3a and 4a with a 50% inhibitory concentration (IC50) value ranging from 0.7 to 2.6 μM. GS-461203 (the active metabolite of sofosbuvir) is not an inhibitor of human DNA and RNA polymerases nor an inhibitor of mitochondrial RNA polymerase.
Antiviral activity: In HCV replicon assays, the effective concentration (EC50) values of sofosbuvir against full-length replicons from genotype 1a, 1b, 2a, 3a and 4a were 0.04, 0.11, 0.05, 0.05 and 0.04 μM, respectively, and EC50 values of sofosbuvir against chimeric 1b replicons encoding NS5B from genotype 2b, 5a or 6a were 0.014 to 0.015 μM. The mean ± SD EC50 of sofosbuvir against chimeric replicons encoding NS5B sequences from clinical isolates was 0.068 ± 0.024 μM for genotype 1a (n = 67), 0.11 ± 0.029 μM for genotype 1b (n = 29), 0.035 ± 0.018 μM for genotype 2 (n = 15) and 0.085 ± 0.034 μM for genotype 3a (n = 106). In these assays, the in vitro antiviral activity of sofosbuvir against the less common genotypes 4, 5 and 6 was similar to that observed for genotypes 1, 2 and 3.
The presence of 40% human serum had no effect on the anti-HCV activity of sofosbuvir.
Resistance: In cell culture: HCV replicons with reduced susceptibility to sofosbuvir have been selected in cell culture for multiple genotypes including 1b, 2a, 2b, 3a, 4a, 5a and 6a. Reduced susceptibility to sofosbuvir was associated with the primary NS5B substitution S282T in all replicon genotypes examined. Site-directed mutagenesis of the S282T substitution in replicons of 8 genotypes conferred 2- to 18-fold reduced susceptibility to sofosbuvir and reduced the replication viral capacity by 89% to 99% compared to the corresponding wild-type. In biochemical assays, recombinant NS5B polymerase from genotypes 1b, 2a, 3a and 4a expressing the S282T substitution showed reduced susceptibility to GS-461203 compared to respective wild-types.
In clinical studies: In a pooled analysis of 991 subjects who received sofosbuvir in Phase 3 studies, 226 subjects qualified for resistance analysis due to virologic failure or early study drug discontinuation and having HCV RNA >1,000 IU/mL. Post-baseline NS5B sequences were available for 225 of the 226 subjects, with deep sequencing data (assay cut off of 1%) from 221 of these subjects. The sofosbuvir-associated resistance substitution S282T was not detected in any of these subjects by deep sequencing or population sequencing. The S282T substitution in NS5B was detected in a single subject receiving SOFOSBUVIR monotherapy in a Phase 2 study. This subject harboured <1% HCV S282T at baseline and developed S282T (>99%) at 4 weeks post-treatment which resulted in a 13.5-fold change in sofosbuvir EC50 and reduced viral replication capacity. The S282T substitution reverted to wild-type over the next 8 weeks and was no longer detectable by deep sequencing at 12 weeks post-treatment.
Two NS5B substitutions, L159F and V321A, were detected in post-treatment relapse samples from multiple genotype 3 HCV infected subjects in the Phase 3 clinical studies. No shift in the phenotypic susceptibility to sofosbuvir or ribavirin of subject isolates with these substitutions was detected. In the study done in subjects with hepatocellular carcinoma awaiting liver transplantation where subjects received up to 48 weeks of sofosbuvir and ribavirin, the L159F substitution emerged in multiple subjects with GT1a or GT2b HCV who experienced virologic failure (breakthrough and relapse). Furthermore, the presence of substitutions L159F and/or C316N at baseline was associated with sofosbuvir breakthrough and relapse post-transplant in multiple subjects infected with GT1b HCV. In addition, S282R and L320F substitutions were detected on treatment by deep sequencing in a pre-transplant subject with GT1a HCV with a partial treatment response. The clinical significance of these findings is unknown.
Effect of baseline HCV polymorphisms on treatment outcome: Baseline NS5B sequences were obtained for 1,292 subjects from Phase 3 studies by population sequencing and the S282T substitution was not detected in any subject with available baseline sequence. In an analysis evaluating the effect of baseline polymorphisms on treatment outcome, no statistically significant association was observed between the presence of any HCV NS5B variant at baseline and treatment outcome.
Cross-resistance: HCV replicons expressing the sofosbuvir-associated resistance substitution S282T were fully susceptible to other classes of anti-HCV agents. Sofosbuvir retained activity against the NS5B substitutions L159F and L320F associated with resistance to other nucleoside inhibitors. Sofosbuvir was fully active against substitutions associated with resistance to other direct-acting antivirals with different mechanisms of actions, such as NS5B non-nucleoside inhibitors, NS3 protease inhibitors and NS5A inhibitors.
Clinical efficacy and safety: The efficacy of sofosbuvir was evaluated in five Phase 3 studies in a total of 1,568 subjects with genotypes 1 to 6 chronic hepatitis C. One study was conducted in treatment-naive subjects with genotype 1, 4, 5 or 6 chronic hepatitis C in combination with peginterferon alfa 2a and ribavirin and the other four studies were conducted in subjects with genotype 2 or 3 chronic hepatitis C in combination with ribavirin including one in treatment-naive subjects, one in interferon intolerant, ineligible or unwilling subjects, one in subjects previously treated with an interferon-based regimen, and one in all subjects irrespective of prior treatment history or ability to receive treatment with interferon. Subjects in these studies had compensated liver disease including cirrhosis. Sofosbuvir was administered at a dose of 400 mg once daily. The ribavirin dose was weight-based at 1,000-1,200 mg daily administered in two divided doses, and the peginterferon alfa 2a dose, where applicable, was 180 μg per week. Treatment duration was fixed in each study and was not guided by subjects' HCV RNA levels (no response guided algorithm).
Plasma HCV RNA values were measured during the clinical studies using the COBAS TaqMan HCV test (version 2.0), for use with the High Pure System. The assay had a lower limit of quantification (LLOQ) of 25 IU/mL. Sustained virologic response (SVR) was the primary endpoint to determine the HCV cure rate for all studies which was defined as HCV RNA less than LLOQ at 12 weeks after the end of treatment (SVR12).
Clinical studies in subjects with genotype 1 and 4 chronic hepatitis C: Treatment-naive subjects - NEUTRINO (study 110): NEUTRINO was an open-label, single-arm study that evaluated 12 weeks of treatment with sofosbuvir in combination with peginterferon alfa 2a and ribavirin in treatment-naive subjects with genotype 1, 4, 5 or 6 HCV infection.
Treated subjects (n = 327) had a median age of 54 years (range: 19 to 70); 64% of the subjects were male; 79% were White; 17% were Black; 14% were Hispanic or Latino; mean body mass index was 29 kg/m2 (range: 18 to 56 kg/m2); 78% had baseline HCV RNA greater than 6 log10 IU/mL; 17% had cirrhosis; 89% had HCV genotype 1 and 11% had HCV genotype 4, 5 or 6.
Table 1 presents the response rates for the treatment group of sofosbuvir + peginterferon alfa + ribavirin. (See Table 1.)
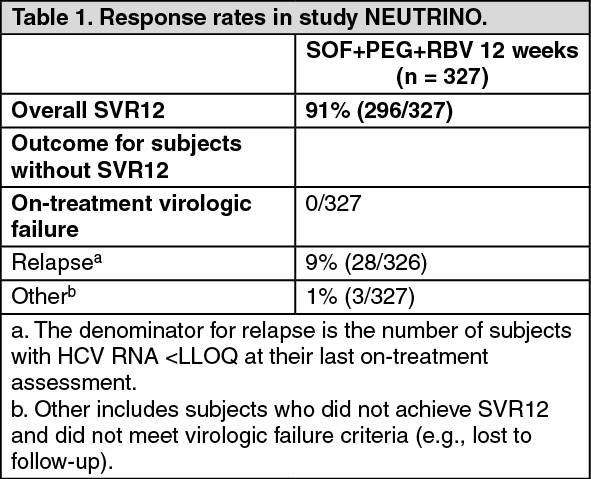 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageResponse rates for selected subgroups are presented in Table 2. (See Table 2.)
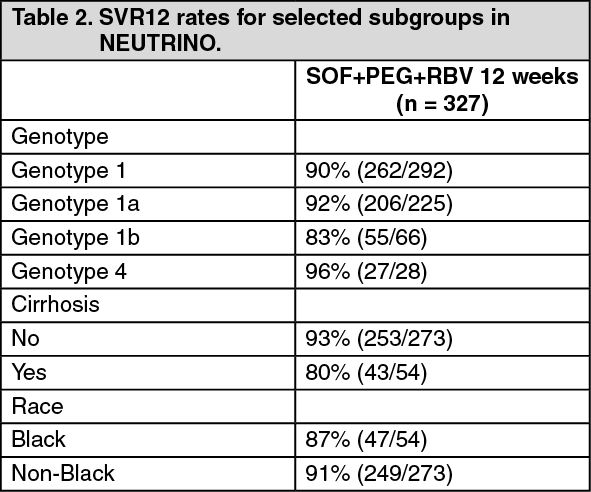 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSVR12 rates were similarly high in subjects with baseline IL28B C/C allele [94/95 (99%)] and non-C/C (C/T or T/T) allele [202/232 (87%)].
27/28 patients with genotype 4 HCV achieved SVR12. A single subject with genotype 5 and all 6 subjects with genotype 6 HCV infection in this study achieved SVR12.
Clinical studies in subjects with genotype 2 and 3 chronic hepatitis C: Treatment-naive adults - FISSION (study 1231): FISSION was a randomised, open-label, active-controlled study that evaluated 12 weeks of treatment with sofosbuvir and ribavirin compared to 24 weeks of treatment with peginterferon alfa 2a and ribavirin in treatment-naive subjects with genotype 2 or 3 HCV infection. The ribavirin doses used in the sofosbuvir + ribavirin and peginterferon alfa 2a + ribavirin arms were weight-based 1,000-1,200 mg/day and 800 mg/day regardless of weight, respectively. Subjects were randomised in a 1:1 ratio and stratified by cirrhosis (presence versus absence), HCV genotype (2 versus 3) and baseline HCV RNA level (<6 log10 IU/mL versus ≥6 log10 IU/mL). Subjects with genotype 2 or 3 HCV were enrolled in an approximately 1:3 ratio.
Treated subjects (n = 499) had a median age of 50 years (range: 19 to 77); 66% of the subjects were male; 87% were White; 3% were Black; 14% were Hispanic or Latino; mean body mass index was 28 kg/m2 (range: 17 to 52 kg/m2); 57% had baseline HCV RNA levels greater than 6 log10 IU/mL; 20% had cirrhosis; 72% had HCV genotype 3.
Table 3 presents the response rates for the treatment groups of sofosbuvir + ribavirin and peginterferon alfa + ribavirin. (See Table 3.)
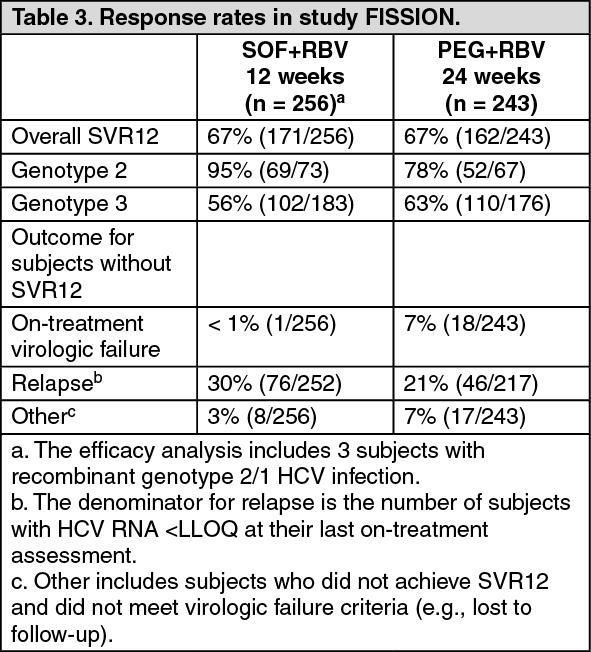 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe difference in the overall SVR12 rates between sofosbuvir + ribavirin and peginterferon alfa + ribavirin treatment groups was 0.3% (95% confidence interval: -7.5% to 8.0%) and the study met the predefined non-inferiority criterion.
Response rates for subjects with cirrhosis at baseline are presented in Table 4 by HCV genotype. (See Table 4.)
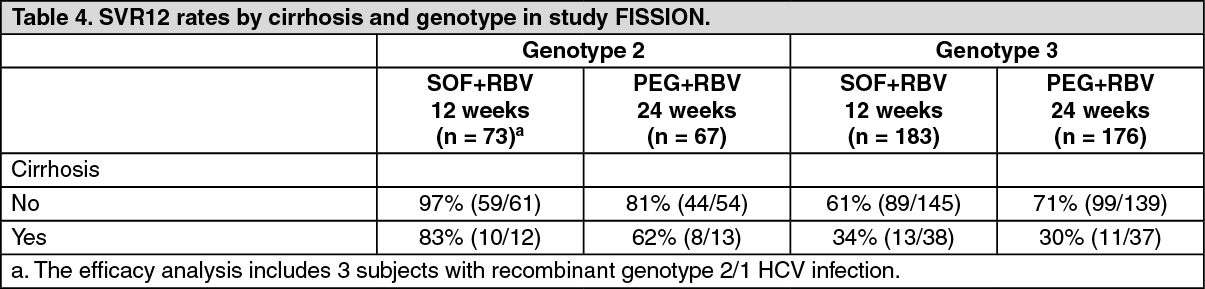 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageInterferon intolerant, ineligible or unwilling adults - POSITRON (study 107): POSITRON was a randomized, double-blinded, placebo-controlled study that evaluated 12 weeks of treatment with sofosbuvir and ribavirin (n = 207) compared to placebo (n = 71) in subjects who are interferon intolerant, ineligible or unwilling. Subjects were randomized in 3:1 ratio and stratified by cirrhosis (presence versus absence).
Treated subjects (n = 278) had a median age of 54 years (range: 21 to 75); 54% of the subjects were male; 91% were White; 5% were Black; 11% were Hispanic or Latino; mean body mass index was 28 kg/m2 (range: 18 to 53 kg/m2); 70% had baseline HCV RNA levels greater than 6 log10 IU/mL; 16% had cirrhosis; 49% had HCV genotype 3. The proportions of subjects who were interferon intolerant, ineligible, or unwilling were 9%, 44%, and 47%, respectively. Most subjects had no prior HCV treatment (81.3%).
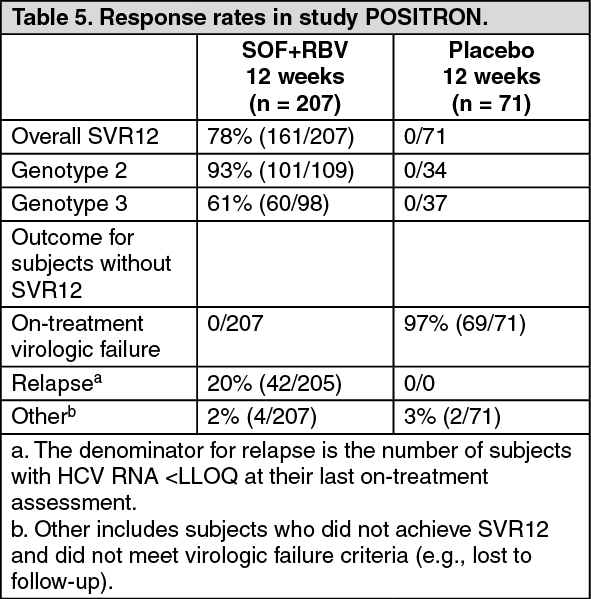
Table 5 presents the response rates for the treatment groups of sofosbuvir + ribavirin and placebo. (See Table 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe SVR12 rate in the sofosbuvir + ribavirin treatment group was statistically significant when compared to placebo (p <0.001).
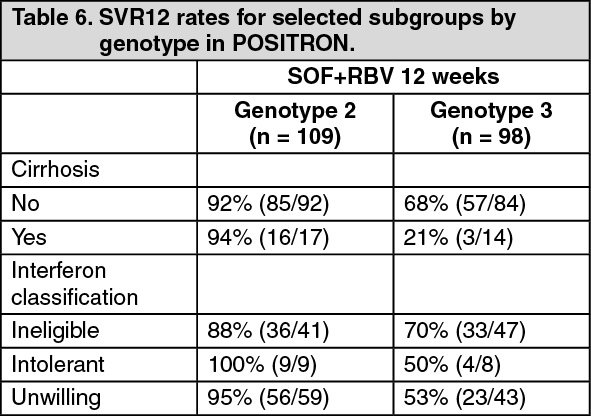
Table 6 presents the subgroup analysis by genotype for cirrhosis and interferon classification. (See Table 6.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePreviously treated adults - FUSION (study 108): FUSION was a randomised, double-blinded study that evaluated 12 or 16 weeks of treatment with sofosbuvir and ribavirin in subjects who did not achieve SVR with prior interferon-based treatment (relapsers and nonresponders). Subjects were randomised in a 1:1 ratio and stratified by cirrhosis (presence versus absence) and HCV genotype (2 versus 3).
Treated subjects (n = 201) had a median age of 56 years (range: 24 to 70); 70% of the subjects were male; 87% were White; 3% were Black; 9% were Hispanic or Latino; mean body mass index was 29 kg/m2 (range: 19 to 44 kg/m2); 73% had baseline HCV RNA levels greater than 6 log10 IU/mL; 34% had cirrhosis; 63% had HCV genotype 3; 75% were prior relapsers.
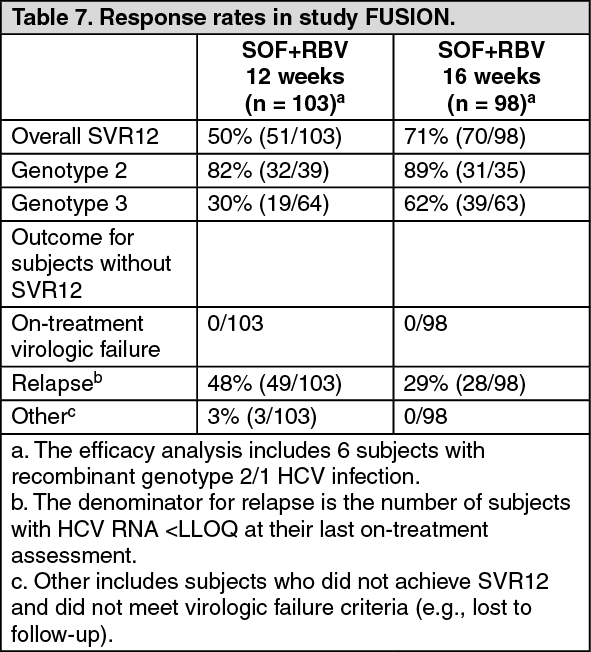
Table 7 presents the response rates for the treatment groups of sofosbuvir + ribavirin for 12 weeks and 16 weeks. (See Table 7.)
 Click on icon to see table/diagram/image
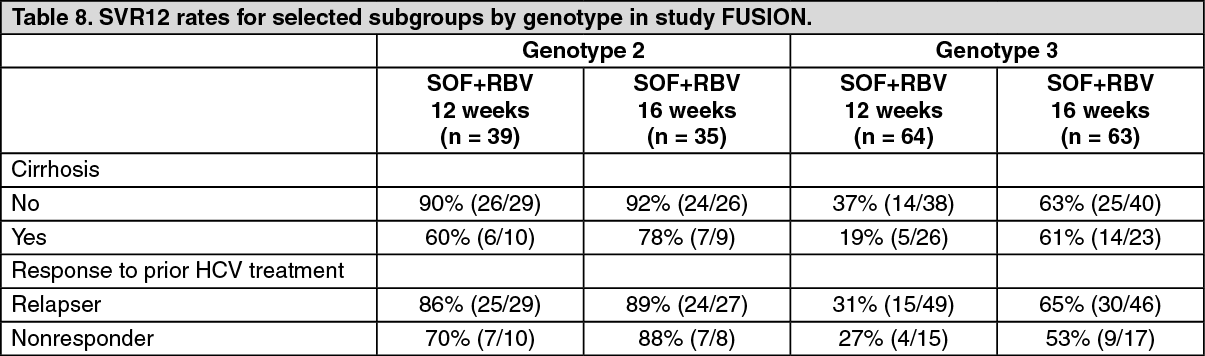
Click on icon to see table/diagram/imageTable 8 presents the subgroup analysis by genotype for cirrhosis and response to prior HCV treatment. (See Table 8.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageTreatment-naive and previously treated adults - VALENCE (study 133): VALENCE was a Phase 3 study that evaluated sofosbuvir in combination with weight-based ribavirin for the treatment of genotype 2 or 3 HCV infection in treatment-naive subjects or subjects who did not achieve SVR with prior interferon-based treatment, including subjects with compensated cirrhosis. The study was designed as a direct comparison of sofosbuvir and ribavirin versus placebo for 12 weeks. However, based on emerging data, the study was unblinded and all HCV genotype 2 subjects continued to receive sofosbuvir and ribavirin for 12 weeks, whilst treatment for HCV genotype 3 subjects was extended to 24 weeks. Eleven HCV genotype 3 subjects had already completed treatment with sofosbuvir and ribavirin for 12 weeks at the time of the amendment.
Treated subjects (n = 419) had a median age of 51 years (range: 19 to 74); 60% of the subjects were male; median body mass index was 25 kg/m2 (range: 17 to 44 kg/m2); the mean baseline HCV RNA level was 6.4 log10 IU/mL; 21% had cirrhosis; 78% had HCV genotype 3; 65% were prior relapsers.
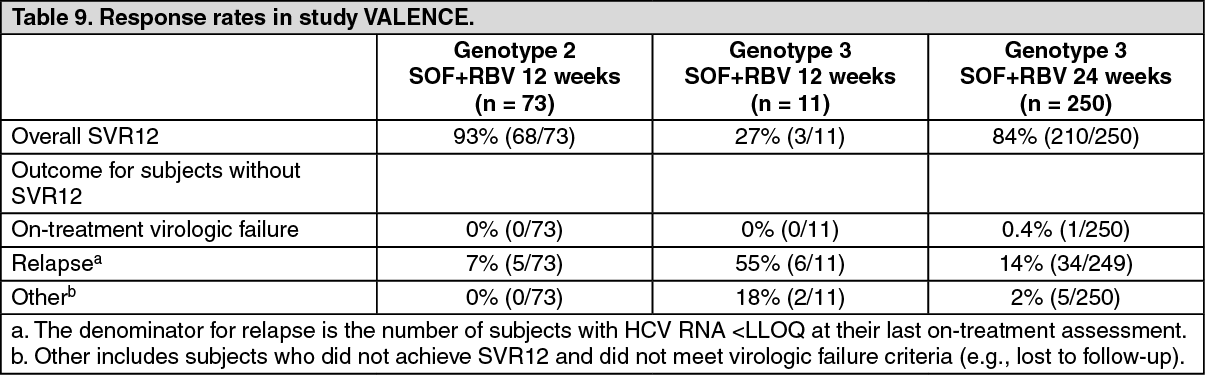
Table 9 presents the response rates for the treatment groups of sofosbuvir + ribavirin for 12 weeks and 24 weeks. Placebo recipients are not included in the tables since none achieved SVR12. (See Table 9.)
 Click on icon to see table/diagram/image
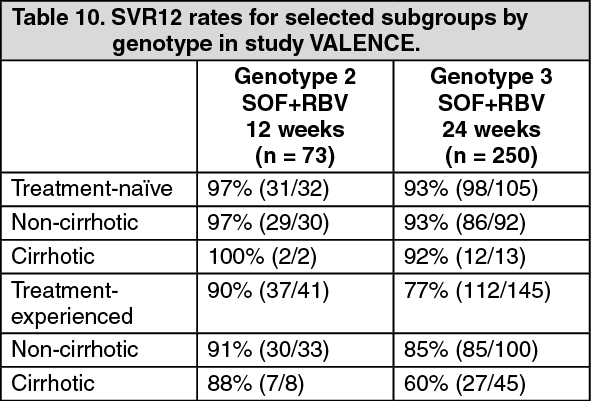
Click on icon to see table/diagram/imageTable 10 presents the subgroup analysis by genotype for cirrhosis and exposure to prior HCV treatment. (See Table 10.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSVR12 to SVR24 concordance: The concordance between SVR12 and SVR24 (SVR 24 weeks after the end of the treatment) following treatment with sofosbuvir in combination with ribavirin or ribavirin and pegylated interferon demonstrates a positive predictive value of 99% and a negative predictive value of 99%.
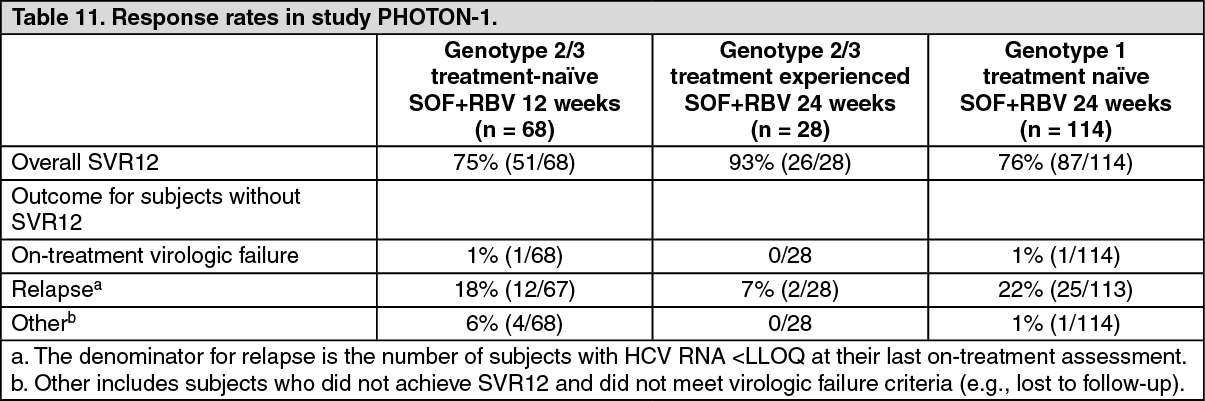
Clinical efficacy and safety in special populations: HCV/HIV co-infected patients - PHOTON-1 (study 123): Sofosbuvir was studied in an open-label clinical study evaluating the safety and efficacy of 12 or 24 weeks of treatment with sofosbuvir and ribavirin in subjects with genotype 1, 2 or 3 chronic hepatitis C co-infected with HIV-1. Genotype 2 and 3 subjects were either treatment-naive or experienced, whereas genotype 1 subjects were naive to prior treatment. Treatment duration was 12 weeks in treatment-naive subjects with genotype 2 or 3 HCV infection, and 24 weeks in treatment-experienced subjects with genotype 3 HCV infection, as well as subjects with genotype 1 HCV infection. Subjects received 400 mg sofosbuvir and weight-based ribavirin (1,000 mg for subjects weighing <75 kg or 1,200 mg for subjects weighing ≥75 kg). Subjects were either not on antiretroviral therapy with a CD4+ cell count >500 cells/mm3 or had virologically suppressed HIV-1 with a CD4+ cell count >200 cells/mm3. 95% of patients received antiretroviral therapy at the time of enrolment. Preliminary SVR12 data are available for 210 subjects.
Table 11 presents the response rates by genotype and exposure to prior HCV treatment. (See Table 11.)
 Click on icon to see table/diagram/image
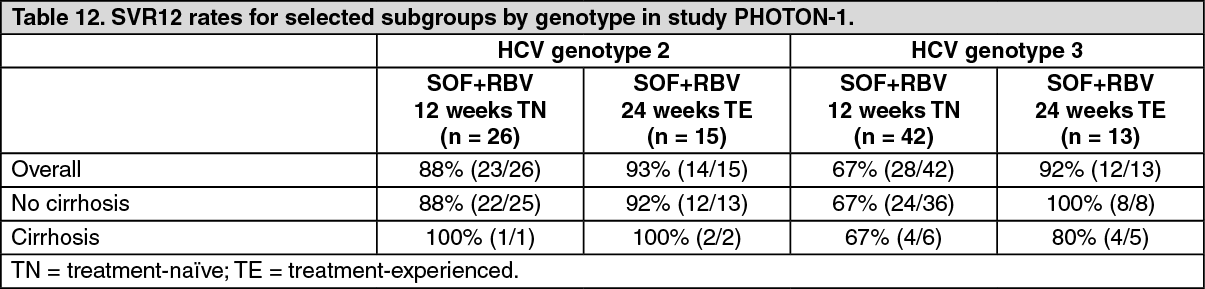
Click on icon to see table/diagram/imageTable 12 presents the subgroup analysis by genotype for cirrhosis. (See Table 12.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn subjects with HCV genotype 1 infection, the SVR rate was 82% (74/90) in subjects with genotype 1a infection and 54% (13/24) in subjects with genotype 1b infection, with relapse accounting for the majority of treatment failures. SVR rates in subjects with HCV genotype 1 infection were 80% (24/30) in subjects with baseline IL28B C/C allele and 75% (62/83) in subjects with baseline IL28B non-C/C alleles.
In the 223 CHC subjects with HIV-1 co-infection, the percentage of CD4+ cells did not change during treatment. Median CD4+ cell count decreases of 85 cells/mm3 and 84 cells/ mm3 were observed at the end of treatment with sofosbuvir +ribavirin for 12 or 24 weeks, respectively. HIV-1 rebound during sofosbuvir +ribavirin treatment occurred in 2 subjects (0.9%) on antiretroviral therapy.
Patients awaiting liver transplantation - Study 2025: Sofosbuvir was studied in HCV infected subjects prior to undergoing liver transplantation in an open-label clinical study evaluating the safety and efficacy of sofosbuvir and ribavirin administered pre-transplant to prevent post-transplant HCV reinfection. The primary endpoint of the study was post-transplant virologic response (pTVR, HCV RNA <LLOQ at 12 weeks post-transplant). HCV infected subjects, regardless of genotype, with hepatocellular carcinoma (HCC) meeting the MILAN criteria received 400 mg sofosbuvir and 1,000-1,200 mg ribavirin daily for a maximum of 24 weeks, subsequently amended to 48 weeks, or until the time of liver transplantation, whichever occurred first. An interim analysis was conducted on 61 subjects who received sofosbuvir and ribavirin; the majority of subjects had HCV genotype 1, 44 subjects were CPT class A and 17 subjects were CPT class B. Of these 61 subjects, 44 subjects underwent liver transplantation following up to 48 weeks of treatment with sofosbuvir and ribavirin; 41 had HCV RNA <LLOQ at the time of transplantation.
The virologic response rates of the 41 subjects transplanted with HCV RNA <LLOQ is described in Table 13. Duration of viral suppression prior to transplantation was the most predictive factor for pTVR in those who were HCV RNA <LLOQ at the time of transplantation. (See Table 13.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageLiver transplant recipients - Study 0126: Sofosbuvir was studied in an open label clinical study evaluating the safety and efficacy of 24 weeks of treatment with sofosbuvir and ribavirin in liver transplant recipients with chronic hepatitis C. Eligible subjects were ≥18 years old and had undergone liver transplantation 6 to 150 months prior to screening. Subjects had HCV RNA ≥104 IU/mL at screening and documented evidence of chronic HCV infection pre-transplantation. The starting dose of ribavirin was 400 mg given in a divided daily dose. If subjects-maintained haemoglobin levels ≥12 g/dL, ribavirin dose was increased at weeks 2, 4, and up to every 4 weeks until the appropriate weight-based dose was reached (1,000 mg daily in subjects <75 kg, 1,200 mg daily in subjects ≥75 kg). The median ribavirin dose was 600 mg 800 mg daily at weeks 4-24.
Forty subjects (33 with HCV genotype 1 infection, 6 with HCV genotype 3 infection, and 1 with HCV genotype 4 infection) were enrolled, 35 of whom had previously failed interferon-based treatment, and 16 of whom had cirrhosis. 28 out of 40 (70%) subjects achieved SVR12: 22/33 (73%) with HCV genotype 1 infection, 6/6 (100%) with HCV genotype 3 infection, and 0/1 (0%) with HCV genotype 4 infection. All subjects who achieved SVR12 achieved SVR24 and SVR48.
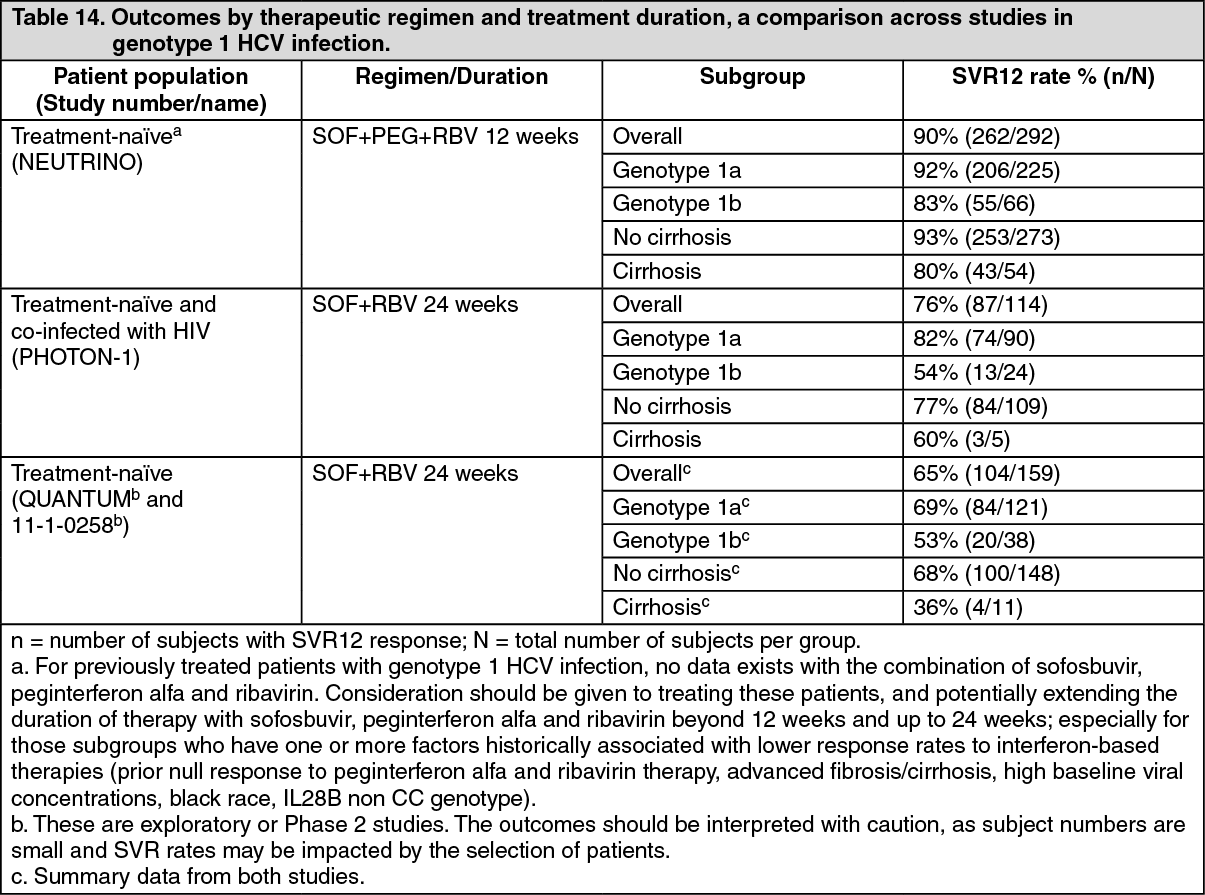
Overview of outcomes by therapeutic regimen and treatment duration, a comparison across studies: The following tables (Table 14 to Table 17) present data from Phase 2 and Phase 3 studies relevant to the dosing to help clinicians determine the best regimen for individual patients. (See Tables 14, 15, 16 and 17.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image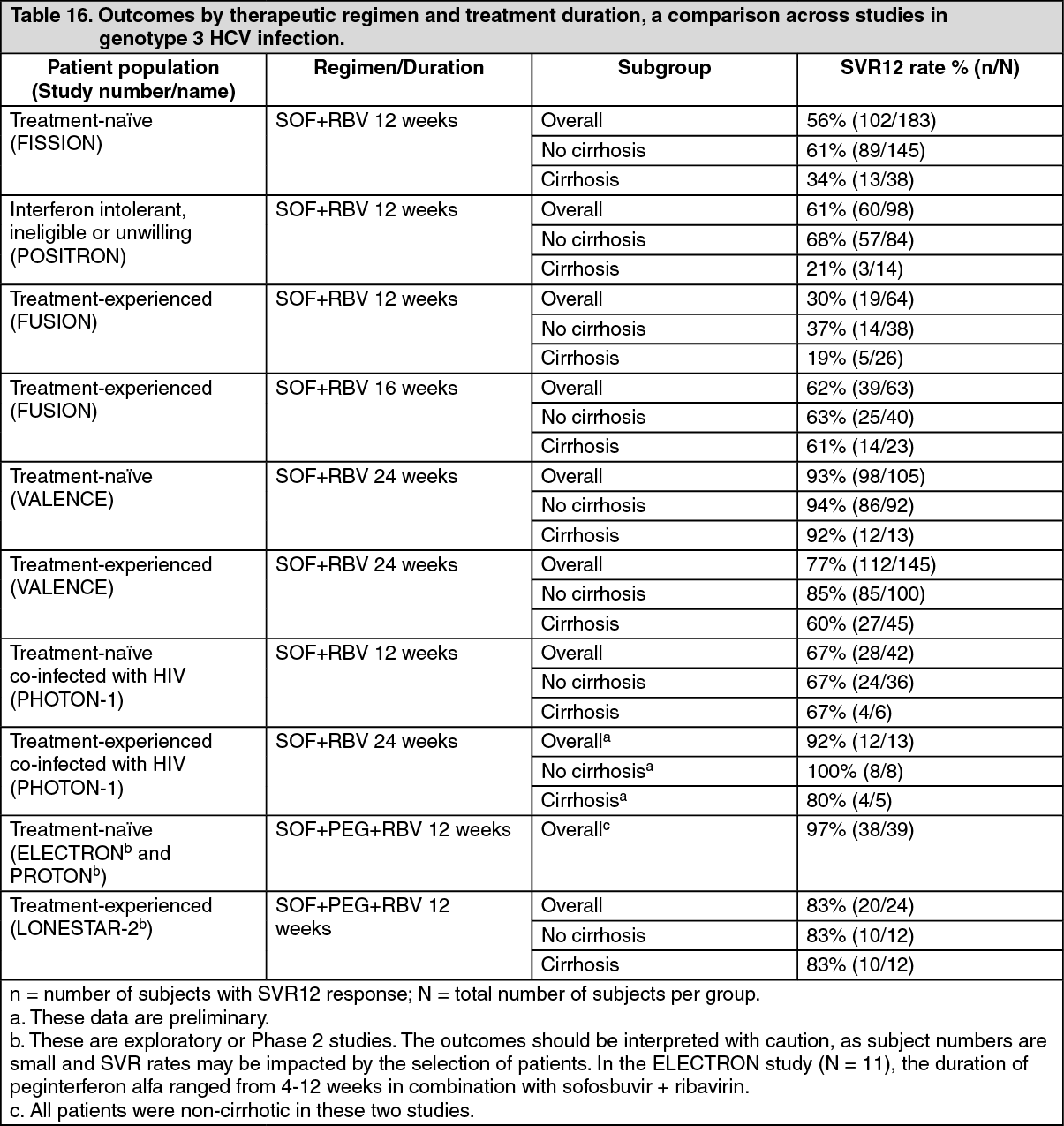 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image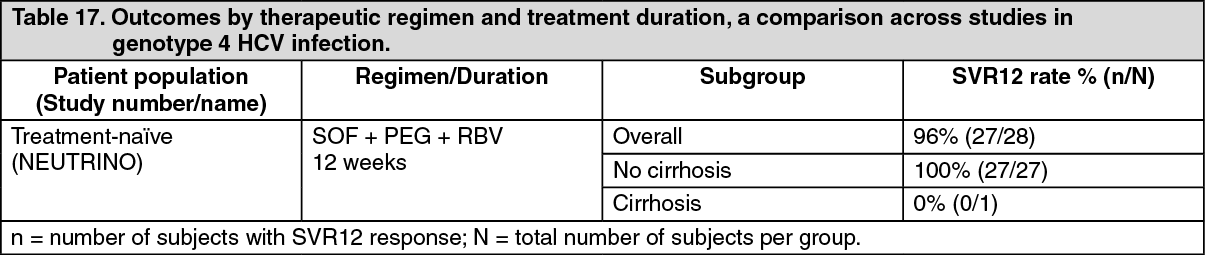 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePharmacokinetics: Sofosbuvir is a nucleotide prodrug that is extensively metabolized. The active metabolite is formed in hepatocytes and not observed in plasma. The predominant (>90%) metabolite, GS-331007, is inactive. It is formed through sequential and parallel pathways to the formation of active metabolite.
Absorption: The pharmacokinetic properties of sofosbuvir and the predominant circulating metabolite GS-331007 have been evaluated. Following oral administration, sofosbuvir was absorbed quickly and the peak plasma concentration was observed, regardless of dose level. Peak plasma concentration of GS-331007 was observed between 2 to 4 hours post-dose.
Effects of food: Relative to fasting conditions, the administration of a single dose of sofosbuvir with a standardized high fat meal slowed the rate of absorption of sofosbuvir. The extent of absorption of sofosbuvir was increased approximately 1.8-fold, with little effect on peak concentration. The exposure to GS-331007 was not altered in the presence of a high-fat meal.
Distribution: Sofosbuvir is not a substrate for hepatic uptake transporters, organic anion-transporting polypeptide (OATP) 1B1 or 1B3, and organic cation transporter (OCT) 1. While subject to active tubular secretion, GS-331007 is not a substrate for renal transporters including organic anion transporter (OAT) 1 or 3, OCT2, MRP2, P-gp, BCRP or MATE1. Sofosbuvir and GS-331007 are not inhibitors of drug transporters P-gp, BCRP, MRP2, BSEP, OATP1B1, OATP1B3 and OCT1. GS-331007 is not an inhibitor of OAT1, OCT2, and MATE1.
Sofosbuvir is approximately 85% bound to human plasma proteins and the binding is independent of drug concentration over the range of 1 μg/mL to 20 μg/mL. Protein binding of GS-331007 was minimal in human plasma.
Biotransformation: Sofosbuvir is extensively metabolized in the liver to form the pharmacologically active nucleoside analog triphosphate GS-461203. The metabolic activation pathway involves sequential hydrolysis of the carboxyl ester moiety catalyzed by human cathepsin A (CatA) or carboxylesterase 1 (CES1) and phosphoramidate cleavage by histidine triad nucleotide-binding protein 1 (HINT1) followed by phosphorylation by the pyrimidine nucleotide biosynthesis pathway. Dephosphorylation results in the formation of nucleoside metabolite GS-331007 that cannot be efficiently rephosphorylated and lacks anti-HCV activity in vitro. Sofosbuvir and GS-331007 are not substrates or inhibitors of UGT1A1 or CYP3A4, CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, and CYP2D6 enzymes.
After a single 400 mg oral dose of [14C]-sofosbuvir, sofosbuvir and GS-331007 accounted for approximately 4% and >90% of drug-related material (sum of molecular weight-adjusted AUC of sofosbuvir and its metabolites) systemic exposure, respectively.
Elimination: Following a single 400 mg oral dose of [14C]-Sofosbuvir, mean total recovery of the dose was greater than 92%, consisting of approximately 80%, 14%, and 2.5% recovered in urine, faeces, and expired air, respectively. The majority of the Sofosbuvir dose recovered in urine was GS-331007 (78%) while 3.5% was recovered as sofosbuvir. This data indicate that renal clearance is the major elimination pathway for GS-331007 with a large part actively secreted. The median terminal half-lives of sofosbuvir and GS-331007 were 0.4 and 27 hours respectively.
Linearity/non-linearity: The dose linearity of sofosbuvir and its primary metabolite, GS-331007, was evaluated in fasted healthy subjects. Sofosbuvir and GS-331007 AUCs are near dose proportional over the dose range of 200 mg to 400 mg.
Pharmacokinetics in special populations: Gender and race: No clinically relevant pharmacokinetic differences due to gender or race have been identified for sofosbuvir and GS- 331007.
Elderly: Population pharmacokinetic analysis in HCV infected subjects showed that within the age range (19 to 75 years) analysed, age did not have a clinically relevant effect on the exposure to sofosbuvir and GS-331007. Clinical studies of sofosbuvir included 65 subjects aged 65 and over. The response rates observed for subjects over 65 years of age were similar to that of younger subjects across treatment groups.
Renal impairment: The pharmacokinetics of sofosbuvir were studied in HCV negative subjects with mild (eGFR ≥50 and <80 mL/min/ 1.73 m2), moderate (eGFR ≥30 and <50 mL/min/1.73 m2), severe renal impairment (eGFR <30 mL/min/1.73 m2) and subjects with ESRD requiring haemodialysis following a single 400 mg dose of sofosbuvir. Relative to subjects with normal renal function (eGFR >80 mL/min/1.73 m2), the sofosbuvir AUC0-inf was 61%, 107% and 171% higher in mild, moderate and severe renal impairment, while the GS-331007 AUC0-inf was 55%, 88% and 451% higher, respectively. In subjects with ESRD, relative to subjects with normal renal function, sofosbuvir AUC0-inf was 28% higher when sofosbuvir was dosed 1 hour before haemodialysis compared with 60% higher when sofosbuvir was dosed 1 hour after haemodialysis. The AUC0-inf of GS-331007 in subjects with ESRD could not be reliably determined. However, data indicate at least 10-fold and 20-fold higher exposure to GS-331007 in ESRD compared to normal subjects when SOFOSBUVIR was administered 1 hour before or 1 hour after haemodialysis, respectively.
Haemodialysis can efficiently remove (53% extraction ratio) the predominant circulating metabolite GS-331007. A 4-hour haemodialysis session removed approximately 18% of administered dose. No dose adjustment is required for patients with mild or moderate renal impairment. The safety of SOFOSBUVIR has not been assessed in patients with severe renal impairment or ESRD (see Precautions).
Hepatic impairment: The pharmacokinetics of sofosbuvir were studied following 7-day dosing of 400 mg sofosbuvir in HCV infected subjects with moderate and severe hepatic impairment (CPT class B and C). Relative to subjects with normal hepatic function, the sofosbuvir AUC0-24 was 126% and 143% higher in moderate and severe hepatic impairment, while the GS-331007 AUC0-24 was 18% and 9% higher, respectively. Population pharmacokinetics analysis in HCV infected subjects indicated that cirrhosis had no clinically relevant effect on the exposure to sofosbuvir and GS-331007. No dose adjustment of sofosbuvir is recommended for patients with mild, moderate and severe hepatic impairment (see Recommended dose under Dosage & Administration and Instruction for Use under Cautions for Usage).
Paediatric population: The pharmacokinetics of sofosbuvir and GS-331007 in paediatric subjects have not been established (see Recommended dose under Dosage & Administration and Instruction for Use under Cautions for Usage).
Pharmacokinetic/pharmacodynamic relationship(s): Efficacy, in terms of rapid virologic response, has been shown to correlate with exposure to sofosbuvir as well as GS- 331007. However, neither of these entities has been evidenced to be a general surrogate marker for efficacy (SVR12) at the therapeutic 400 mg dose.
Toxicology: Preclinical safety data: Not Applicable.