


 Sign Out
Sign Out


Pharmacology: Pharmacodynamics: Mechanism of action: Semaglutide is a GLP-1 analogue with 94% sequence homology to human GLP-1. Semaglutide acts as a GLP-1 receptor agonist that selectively binds to and activates the GLP-1 receptor, the target for native GLP-1.
GLP-1 is a physiological hormone that has multiple actions in glucose and appetite regulation, and in the cardiovascular system. The glucose and appetite effects are specifically mediated via GLP-1 receptors in the pancreas and the brain.
Semaglutide reduces blood glucose in a glucose-dependent manner by stimulating insulin secretion and lowering glucagon secretion when blood glucose is high. The mechanism of blood glucose lowering also involves a minor delay in gastric emptying in the early postprandial phase. During hypoglycaemia, semaglutide diminishes insulin secretion and does not impair glucagon secretion. The mechanism of semaglutide is independent of the route of administration.
Semaglutide reduces body weight and body fat mass through lowered energy intake, involving an overall reduced appetite. In addition, semaglutide reduces the preference for high fat foods.
GLP-1 receptors are expressed in the heart, vasculature, immune system and kidneys. Semaglutide has a beneficial effect on plasma lipids, lowers systolic blood pressure and reduces inflammation in clinical studies. In animal studies, semaglutide attenuates the development of atherosclerosis by preventing aortic plaque progression and reducing inflammation in the plaque.
Pharmacodynamic effects: The pharmacodynamic evaluations described as follows were performed with orally administered semaglutide after 12 weeks of treatment.
Fasting and postprandial glucose: Semaglutide reduces fasting and postprandial glucose concentrations. In patients with type 2 diabetes, treatment with semaglutide resulted in a relative reduction compared to placebo of 22% [13; 30] for fasting glucose and 29% [19; 37] for postprandial glucose.
Glucagon secretion: Semaglutide lowers the postprandial glucagon concentrations. In patients with type 2 diabetes, semaglutide resulted in the following relative reductions in glucagon compared to placebo: postprandial glucagon response of 29% [15; 41].
Gastric emptying: Semaglutide causes a minor delay in early postprandial gastric emptying, with paracetamol exposure (AUC0-1h) 31% [13; 46] lower in the first hour after the meal, thereby reducing the rate at which glucose appears in the circulation postprandially.
Fasting and postprandial lipids: Semaglutide compared to placebo lowered fasting triglyceride and very-low-density lipoproteins (VLDL) cholesterol concentrations by 19% [8; 28] and 20% [5; 33], respectively. The postprandial triglyceride and VLDL cholesterol response to a high fat meal was reduced by 24% [9; 36] and 21% [7; 32], respectively. ApoB48 was reduced both in fasting and postprandial state by 25% [2; 42] and 30% [15; 43], respectively.
Clinical efficacy and safety: The efficacy and safety of Rybelsus have been evaluated in eight global randomised controlled phase 3a trials. In seven trials, the primary objective was the assessment of the glycaemic efficacy; in one trial, the primary objective was the assessment of cardiovascular outcomes.
The trials included 8,842 randomised patients with type 2 diabetes (5,169 treated with semaglutide), including 1,165 patients with moderate renal impairment. Patients had an average age of 61 years (range 18 to 92 years), with 40% of patients ≥ 65 years of age and 8% ≥ 75 years of age. The efficacy of semaglutide was compared with placebo or active controls (sitagliptin, empagliflozin and liraglutide).
The efficacy of semaglutide was not impacted by baseline age, gender, race, ethnicity, body weight, BMI, diabetes duration, upper gastrointestinal disease and level of renal function.
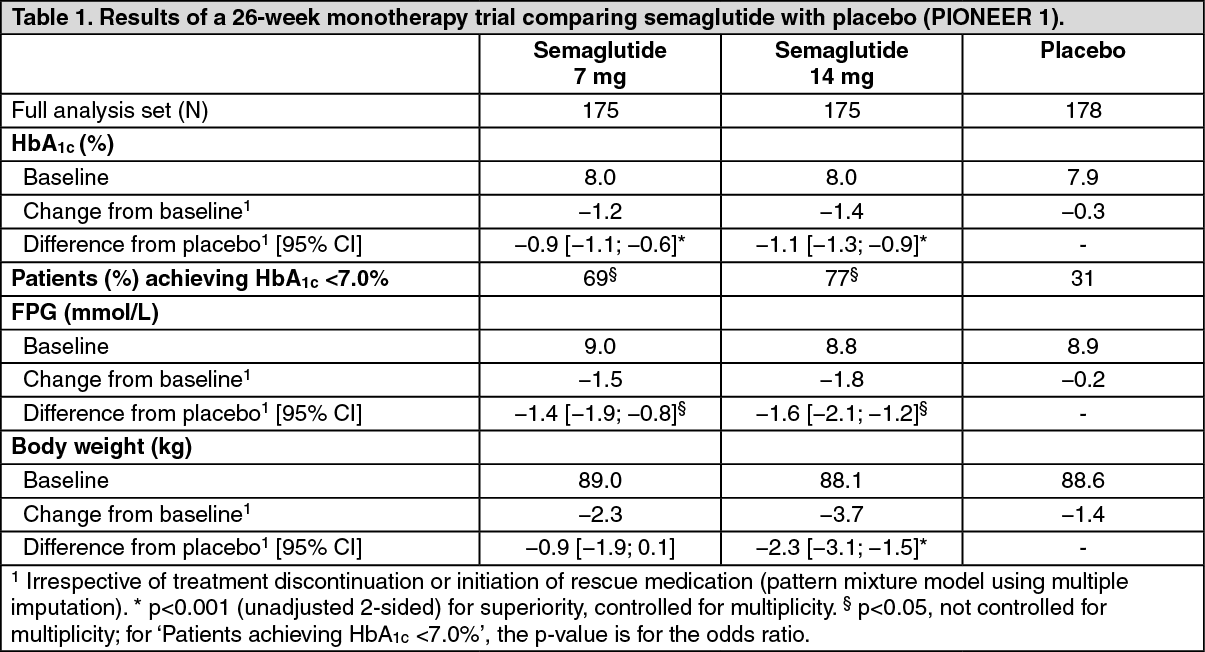
PIONEER 1 - Monotherapy: In a 26-week double-blind trial, 703 patients with type 2 diabetes inadequately controlled with diet and exercise were randomised to semaglutide 3 mg, semaglutide 7 mg, semaglutide 14 mg or placebo once daily. (See Table 1.)
 Click on icon to see table/diagram/image
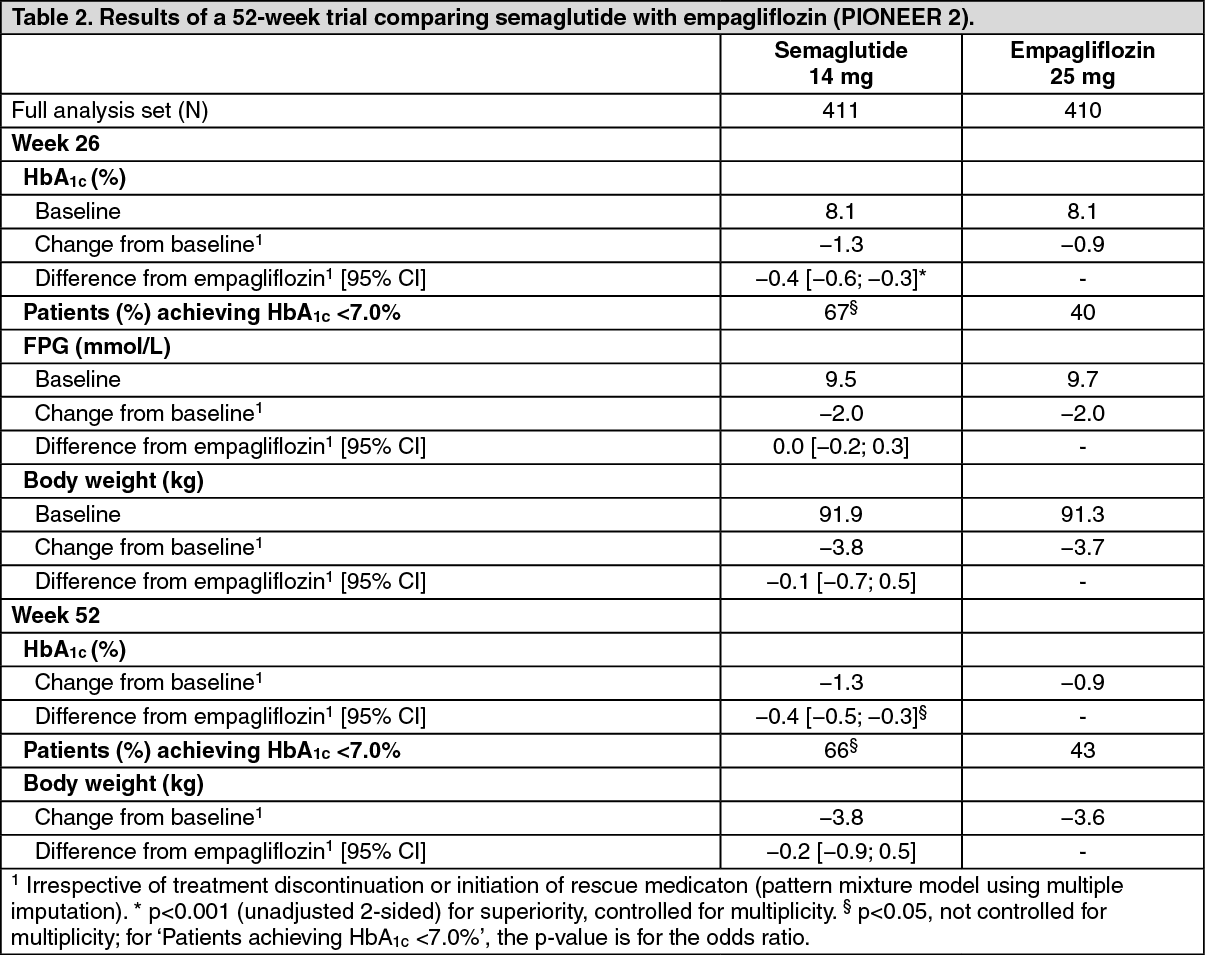
Click on icon to see table/diagram/imagePIONEER 2 - Semaglutide vs. empagliflozin, both in combination with metformin: In a 52-week open-label trial, 822 patients with type 2 diabetes were randomised to semaglutide 14 mg once daily or empagliflozin 25 mg once daily, both in combination with metformin. (See Table 2.)
 Click on icon to see table/diagram/image
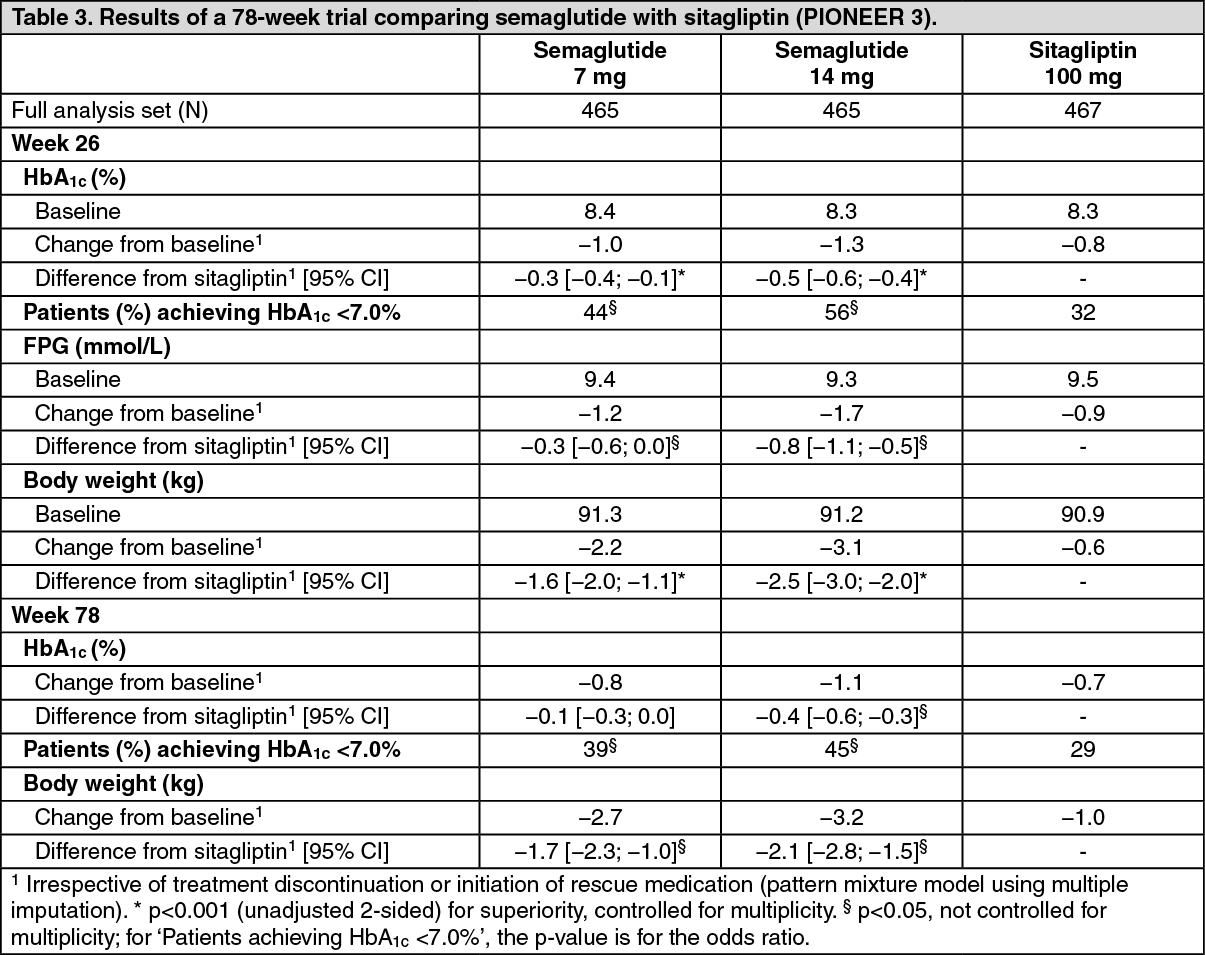
Click on icon to see table/diagram/imagePIONEER 3 - Semaglutide vs. sitagliptin, both in combination with metformin or metformin with sulfonylurea: In a 78-week, double-blind, double-dummy trial, 1,864 patients with type 2 diabetes were randomised to semaglutide 3 mg, semaglutide 7 mg, semaglutide 14 mg or sitagliptin 100 mg once daily, all in combination with metformin alone or metformin and sulfonylurea. Reductions in HbA1c and body weight were sustained throughout the trial duration of 78 weeks. (See Table 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePIONEER 4 - Semaglutide vs. liraglutide and placebo, all in combination with metformin or metformin with an SGLT2 inhibitor: In a 52-week double-blind, double-dummy trial, 711 patients with type 2 diabetes were randomised to semaglutide 14 mg, liraglutide 1.8 mg s.c. injection or placebo once daily, all in combination with metformin or metformin and an SGLT2 inhibitor. (See Table 4.)
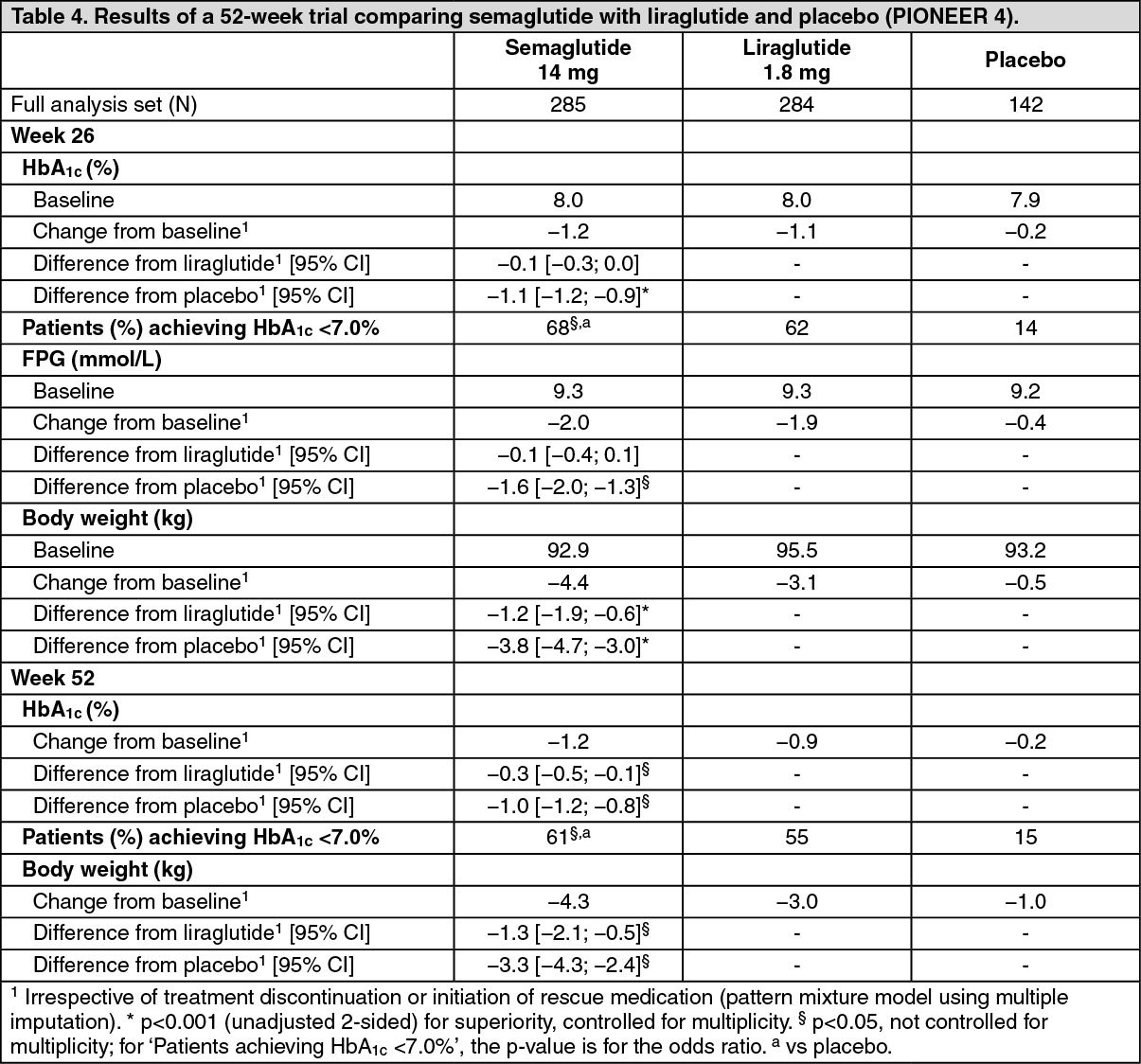 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePIONEER 5 - Semaglutide vs. placebo, both in combination with basal insulin alone, metformin and basal insulin or metformin and/or sulfonylurea, in patients with moderate renal impairment: In a 26-week double-blind trial, 324 patients with type 2 diabetes and moderate renal impairment (eGFR 30-59 ml/min/1.73 m2) were randomised to semaglutide 14 mg or placebo once daily. Trial product was added to the patient's stable pre-trial antidiabetic regimen. (See Table 5.)
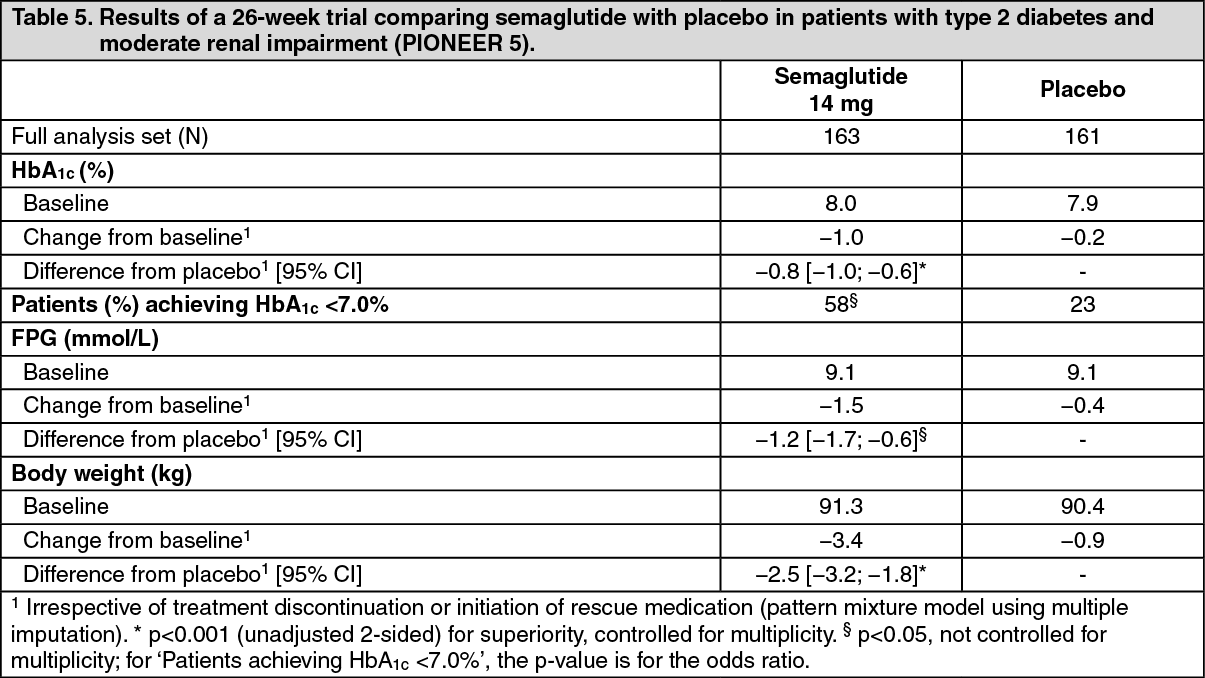 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePIONEER 7 - Semaglutide vs. sitagliptin, both in combination with metformin, SGLT2 inhibitors, sulfonylurea or thiazolidinediones. Flexible-dose-adjustment trial: In a 52-week open-label trial, 504 patients with type 2 diabetes were randomised to semaglutide (flexible dose adjustment of 3 mg, 7 mg, and 14 mg once daily) or sitagliptin 100 mg once daily, all in combination with 1-2 oral glucose-lowering medicinal products (metformin, SGLT2 inhibitors, sulfonylurea or thiazolidinediones). The dose of semaglutide was adjusted every 8 weeks based on patient's glycaemic response and tolerability. The sitagliptin 100 mg dose was fixed. The efficacy and safety of semaglutide were evaluated at week 52.
At week 52, the proportion of patients on treatment with semaglutide 3 mg, 7 mg and 14 mg was approximately 10%, 30% and 60%, respectively. (See Table 6.)
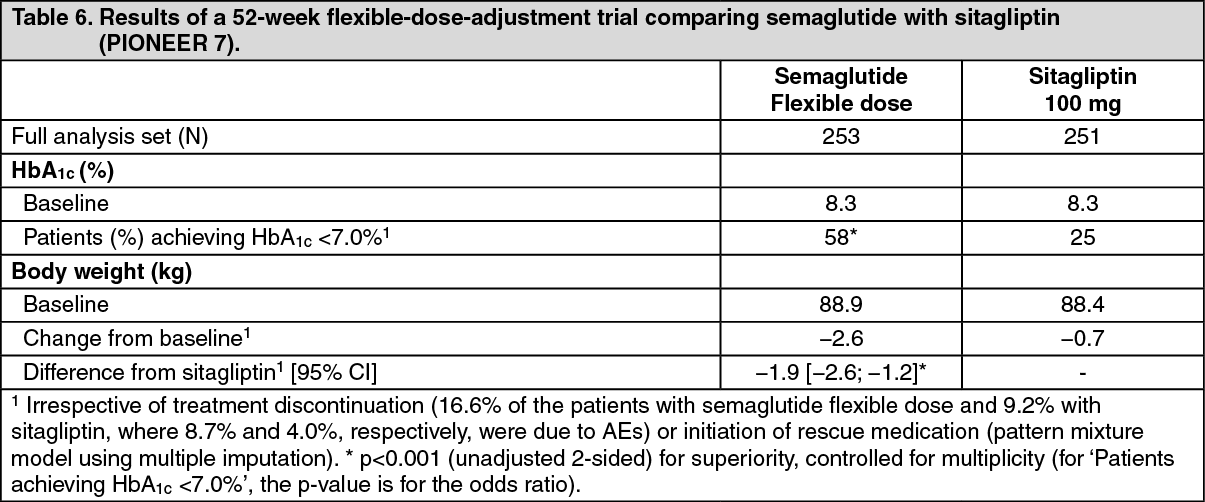 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePIONEER 8 - Semaglutide vs. placebo, both in combination with insulin with or without metformin: In a 52-week double-blind trial, 731 patients with type 2 diabetes inadequately controlled on insulin (basal, basal/bolus or premixed) with or without metformin were randomised to semaglutide 3 mg, semaglutide 7 mg, semaglutide 14 mg or placebo once daily. (See Table 7.)
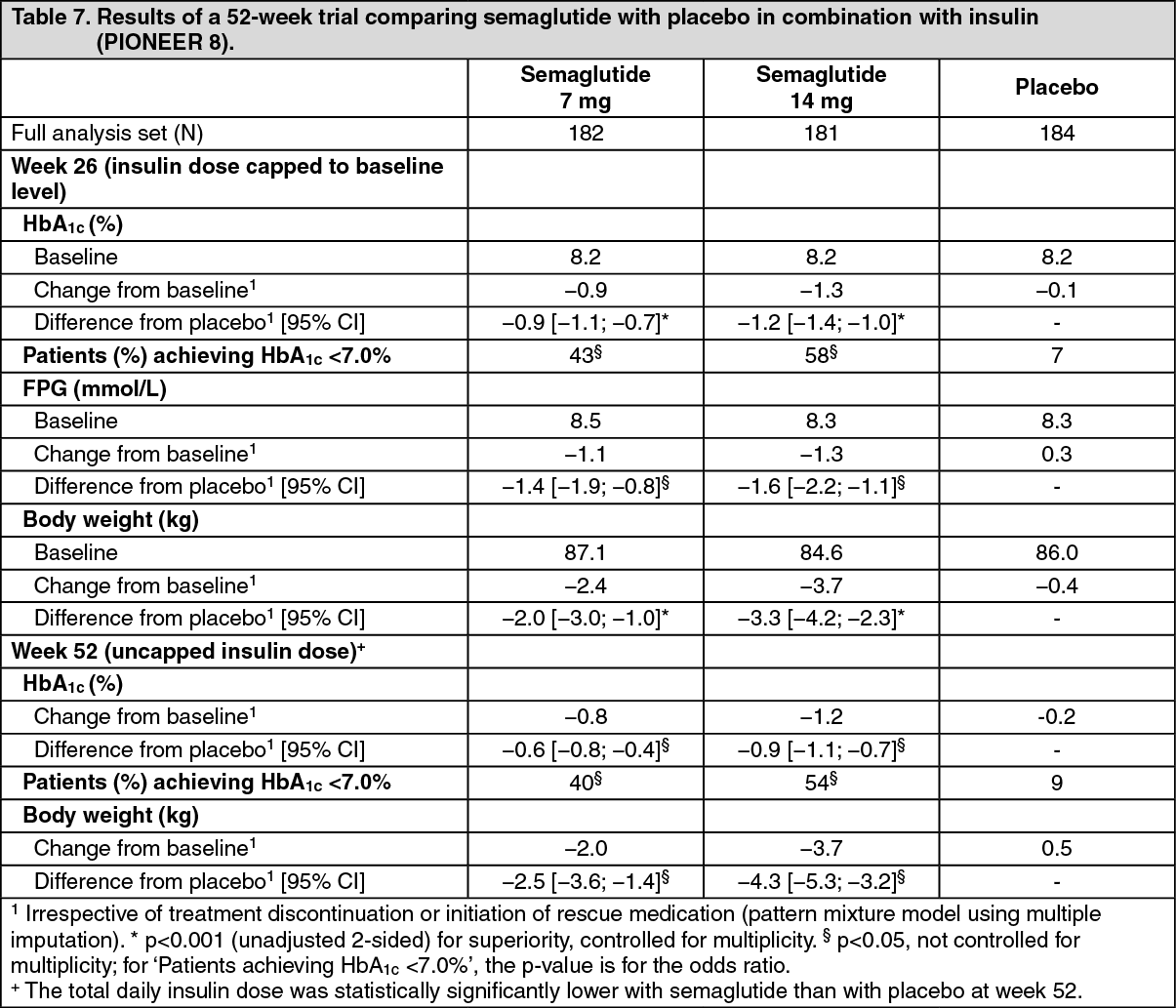 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageCardiovascular evaluation: In a double-blind trial (PIONEER 6), 3,183 patients with type 2 diabetes at high cardiovascular risk were randomised to Rybelsus 14 mg once daily or placebo in addition to standard-of-care. The median observation period was 16 months.
The primary endpoint was time from randomisation to first occurrence of a major adverse cardiovascular event (MACE): cardiovascular death, non-fatal myocardial infarction or non-fatal stroke.
Patients eligible to enter the trial were: 50 years of age or older and with established cardiovascular disease and/or chronic kidney disease, or 60 years of age or older and with cardiovascular risk factors only. In total, 1,797 patients (56.5%) had established cardiovascular disease without chronic kidney disease, 354 (11.1%) had chronic kidney disease only and 544 (17.1%) had both cardiovascular disease and kidney disease. 488 patients (15.3%) had cardiovascular risk factors only. The mean age at baseline was 66 years, and 68% of the patients were men. The mean duration of diabetes was 14.9 years and the mean BMI was 32.3 kg/m2. Medical history included stroke (11.7%) and myocardial infarction (36.1%).
The total number of first MACE was 137: 61 (3.8%) with semaglutide and 76 (4.8%) with placebo. The analysis of time to first MACE resulted in a HR of 0.79 [0.57; 1.11]95% CI. (See Figure 1.)
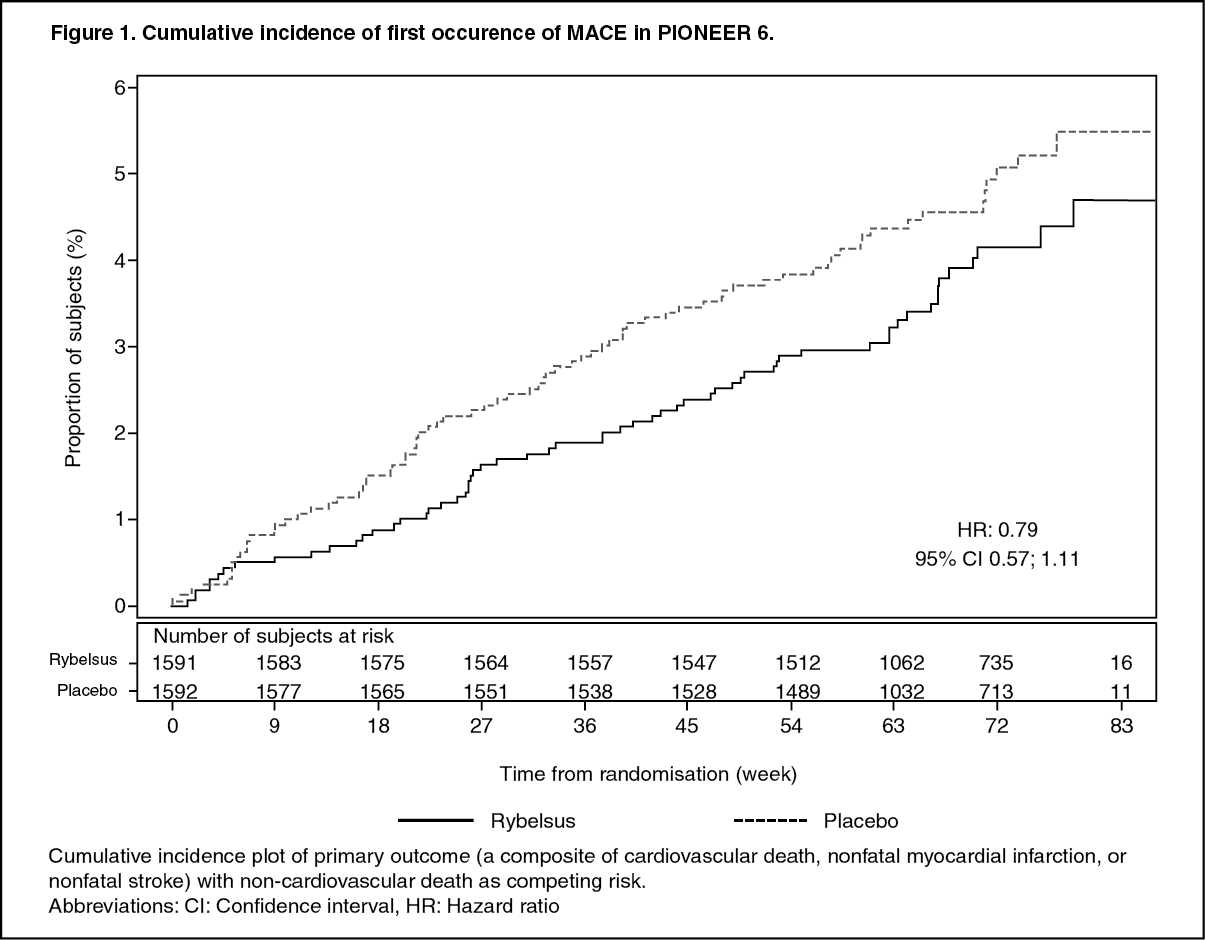 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe treatment effect for the primary composite endpoint and its components in the PIONEER 6 trial is shown in Figure 2. (See Figure 2.)
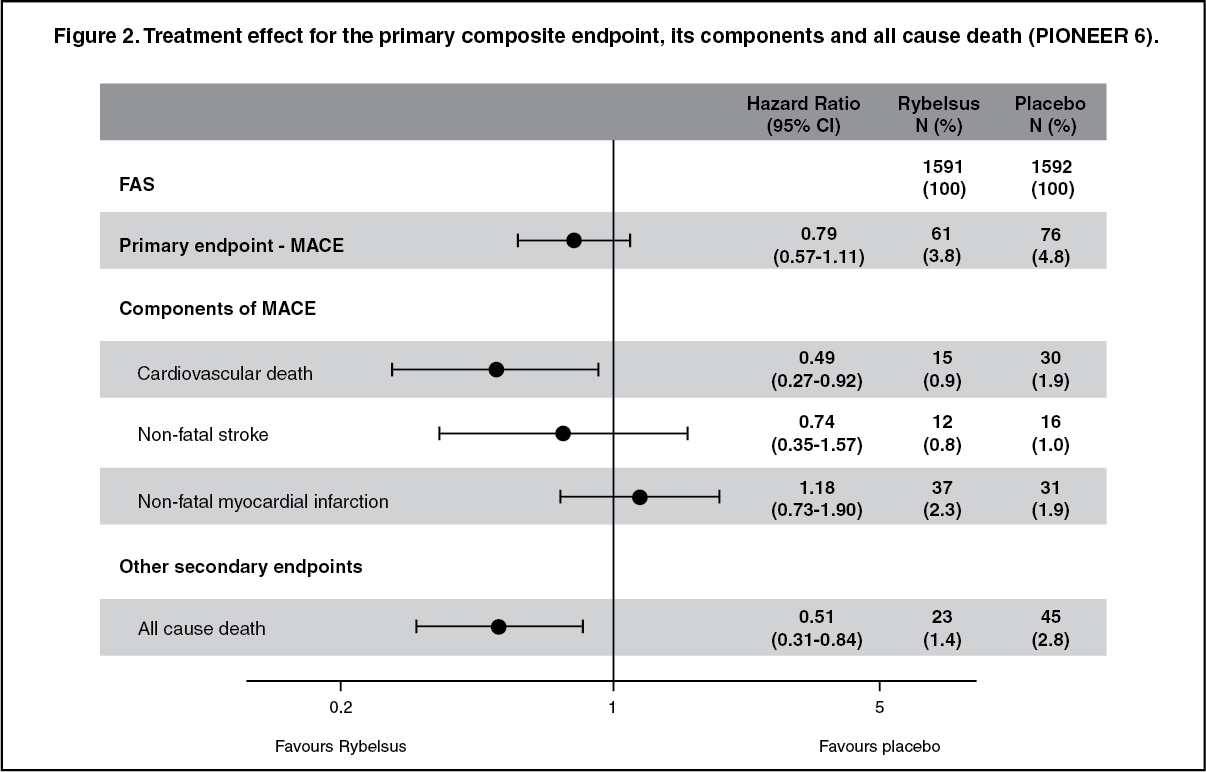 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageBody weight: By end-of-treatment, 27-45% of the patients had achieved a weight loss of ≥5% and 6-16% had achieved a weight loss of ≥10% with semaglutide, compared with 12-39% and 2-8%, respectively, with the active comparators.
Blood pressure: Treatment with semaglutide had reduced systolic blood pressure by 2-7 mmHg.
Pharmacokinetics: Orally administered semaglutide has a low absolute bioavailability and a variable absorption. Daily administration according to the recommended posology in combination with a long half-life reduces day-to-day fluctuation of the exposure.
The pharmacokinetics of semaglutide have been extensively characterised in healthy subjects and patients with type 2 diabetes. Following oral administration, maximum plasma concentration of semaglutide occurred 1 hour post dose. Steady-state exposure was reached after 4-5 weeks of once-daily administration. In patients with type 2 diabetes, the average steady-state concentrations were approximately 6.7 nmol/L and 14.6 nmol/L with semaglutide 7 mg and 14 mg, respectively; with 90% of subjects treated with semaglutide 7 mg having an average concentration between 1.7 and 22.7 nmol/L and 90% of subjects treated with semaglutide 14 mg having an average concentration between 3.7 and 41.3 nmol/L. Systemic exposure of semaglutide increased in a dose-proportional manner.
Based on in vitro data, salcaprozate sodium facilitates absorption of semaglutide. The absorption of semaglutide predominantly occurs in the stomach.
The estimated bioavailability of semaglutide is approximately 1% following oral administration.
The between-subject variability in absorption was high (coefficient of variation was approximately 100%). The estimation of the within-subject variability in bioavailability was not reliable.
Absorption of semaglutide is decreased if taken with food or large volumes of water. A longer post-dose fasting period results in higher absorption.
Distribution: The estimated absolute volume of distribution is approximately 8 L in subjects with type 2 diabetes. Semaglutide is extensively bound to plasma proteins (>99%).
Biotransformation: Semaglutide is metabolised through proteolytic cleavage of the peptide backbone and sequential beta-oxidation of the fatty acid sidechain. The enzyme neutral endopeptidase (NEP) is expected to be involved in the metabolism of semaglutide.
Elimination: The primary excretion routes of semaglutide-related material are via the urine and faeces.
Approximately 3% of the absorbed dose is excreted as intact semaglutide via the urine.
With an elimination half-life of approximately 1 week, semaglutide will be present in the circulation for about 5 weeks after the last dose. The clearance of semaglutide in patients with type 2 diabetes is approximately 0.04 L/h.
Switching between oral and subcutaneous (s.c.) administration: The effect of switching between oral and s.c. semaglutide cannot easily be predicted because of the high pharmacokinetic variability of oral semaglutide. Exposure after oral semaglutide 14 mg once daily is comparable to s.c. semaglutide 0.5 mg once weekly. An oral dose equivalent to 1.0 mg of s.c. semaglutide has not been established.
Special populations: Elderly: Age had no effect on the pharmacokinetics of semaglutide based on data from clinical trials, which studied patients up to 92 years of age.
Gender: Gender had no clinically meaningful effects on the pharmacokinetics of semaglutide.
Race and ethnicity: Race (White, Black or African-American, Asian) and ethnicity (Hispanic or Latino, not Hispanic or Latino) had no effect on the pharmacokinetics of semaglutide.
Body weight: Body weight had an effect on the exposure of semaglutide. Higher body weight was associated with lower exposure. Semaglutide provided adequate systemic exposure over the body weight range of 40-188 kg evaluated in the clinical trials.
Renal impairment: Renal impairment did not impact the pharmacokinetics of semaglutide in a clinically relevant manner. The pharmacokinetics of semaglutide were evaluated in patients with mild, moderate or severe renal impairment and patients with end-stage renal disease on dialysis compared with subjects with normal renal function in a study with 10 consecutive days of once-daily doses of semaglutide. This was also shown for subjects with type 2 diabetes and renal impairment based on data from phase 3a studies.
Hepatic impairment: Hepatic impairment did not impact the pharmacokinetics of semaglutide in a clinically relevant manner. The pharmacokinetics of semaglutide were evaluated in patients with mild, moderate or severe hepatic impairment compared with subjects with normal hepatic function in a study with 10 consecutive days of once-daily doses of semaglutide.
Upper GI tract disease: Upper GI tract disease (chronic gastritis and/or gastroesophageal reflux disease) did not impact the pharmacokinetics of semaglutide in a clinically relevant manner. The pharmacokinetics were evaluated in patients with type 2 diabetes with or without upper GI tract disease dosed for 10 consecutive days with once-daily doses of semaglutide. This was also shown for subjects with type 2 diabetes and upper GI tract disease based on data from phase 3a studies.
Paediatric population: Semaglutide has not been studied in paediatric patients.
Toxicology: Preclinical safety data: Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity or genotoxicity.
Non-lethal thyroid C-cell tumours observed in rodents are a class effect for GLP-1 receptor agonists. In 2-year carcinogenicity studies in rats and mice, semaglutide caused thyroid C-cell tumours at clinically relevant exposures. No other treatment-related tumours were observed. The rodent C-cell tumours are caused by a non-genotoxic, specific GLP-1 receptor mediated mechanism to which rodents are particularly sensitive. The relevance for humans is considered to be low, but cannot be completely excluded.
In fertility studies in rats, semaglutide did not affect mating performance or male fertility. In female rats, an increase in oestrous cycle length and a small reduction in corpora lutea (ovulations) were observed at doses associated with maternal body weight loss.
In embryo-foetal development studies in rats, semaglutide caused embryotoxicity below clinically relevant exposures. Semaglutide caused marked reductions in maternal body weight and reductions in embryonic survival and growth. In foetuses, major skeletal and visceral malformations were observed, including effects on long bones, ribs, vertebrae, tail, blood vessels and brain ventricles. Mechanistic evaluations indicated that the embryotoxicity involved a GLP-1 receptor mediated impairment of the nutrient supply to the embryo across the rat yolk sac. Due to species differences in yolk sac anatomy and function, and due to the lack of GLP-1 receptor expression in the yolk sac of non-human primates, this mechanism is considered unlikely to be of relevance to humans. However, a direct effect of semaglutide on the foetus cannot be excluded.
In developmental toxicity studies in rabbits and cynomolgus monkeys, increased pregnancy loss and slightly increased incidence of foetal abnormalities were observed at clinically relevant exposures. The findings coincided with marked maternal body weight loss of up to 16%. Whether these effects are related to the decreased maternal food consumption as a direct GLP-1 effect is unknown.
Postnatal growth and development were evaluated in cynomolgus monkeys. Infants were slightly smaller at delivery, but recovered during the lactation period.
In juvenile rats, semaglutide caused delayed sexual maturation in both males and females. These delays had no impact upon fertility and reproductive capacity of either sex, or on the ability of the females to maintain pregnancy.