Pharmacotherapeutic group: Sex hormones and modulators of the genital systems, gonadotropins.
ATC code: G03GA10.
Pharmacology: Pharmacodynamics: Mechanism of action: The most important effect resulting from parenteral administration of FSH is the development of multiple mature follicles.
Follitropin delta is a recombinant human FSH. The amino acid sequences of the two FSH subunits in follitropin delta are identical to the endogenous human FSH sequences.
The expressing cell line can influence the characteristics of the recombinant FSH, and differences in glycosylation profile, sialic acid pattern and isoform profile have been documented between follitropin delta produced in a human cell line and recombinant FSH products such as follitropin alfa and follitropin beta produced in Chinese hamster ovary (CHO) cell lines. The glycosylation of FSH in follitropin delta contains both α2,3 and α2,6-linked sialic acid (2,6-linked sialic acid is absent in CHO-derived recombinant FSH), different sugars such as N-acetylgalactosamine, carries additional linkages between carbohydrates such as bisecting N-acetylglucosamine and antennary fucose, and has a higher proportion of tetra-antennary structures and higher overall sialic acid content than CHO-derived recombinant FSH.
Pharmacodynamic effects: Comparisons of REKOVELLE versus follitropin alfa indicate that the differences in glycosylation influence both the pharmacokinetic and pharmacodynamic profile. Following daily administration of equal IU doses of REKOVELLE and follitropin alfa as determined in the rat in vivo bioassay (Steelman-Pohley assay), higher ovarian response (i.e. estradiol, inhibin B and follicular volume) was observed in patients after administration of REKOVELLE compared to follitropin alfa. As the Steelman-Pohley bioassay might not fully reflect the potency of the FSH in REKOVELLE in humans, REKOVELLE is dosed in micrograms and not in IU. Furthermore, 11 to 27% lower microgram doses of REKOVELLE were sufficient to obtain the same pharmacodynamic response as 11 micrograms (150 IU) filled-by-mass follitropin alfa in terms of follicular development and related hormones. Consequently, the recommended REKOVELLE doses in micrograms are not applicable to other recombinant FSH preparations.
The number of oocytes retrieved increases with the dose of REKOVELLE and serum AMH concentration. Conversely, increasing body weight leads to a decrease in the number of oocytes retrieved (only clinically relevant for REKOVELLE doses below 12 micrograms). Consequently, the REKOVELLE dosing regimen is based on serum AMH concentration and furthermore on body weight for doses lower than 12 micrograms (see Dosage & Administration).
Clinical efficacy and safety: The ESTHER-1 trial was a randomized, assessor-blinded, controlled trial in 1,326 IVF/ICSI patients. The trial compared the individualized dosing regimen of REKOVELLE where the daily dose is established for each patient and fixed throughout stimulation with no adjustments (see Dosage & Administration) to follitropin alfa filled-by-mass at a starting dose of 11 micrograms (150 IU) for the first five days followed by dose adjustments from day 6 of stimulation based on follicular development. The patients were up to 40 years of age and had regular menstrual cycles presumed to be ovulatory. Single blastocyst transfer on day 5 was compulsory with the exception of patients 38-40 years in whom double blastocyst transfer was performed if no good-quality blastocysts were available. The two co-primary endpoints were ongoing pregnancy rate and ongoing implantation rate in the fresh cycle, defined as at least one intrauterine viable fetus 10-11 weeks after transfer and number of intrauterine viable fetuses 10-11 weeks after transfer divided by number of blastocysts transferred, respectively.
The trial demonstrated that REKOVELLE was at least as effective as follitropin alfa in terms of ongoing pregnancy rate and ongoing implantation rate, as shown in Table 1. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The clinical value of the AMH-based dosing regimen of REKOVELLE was also assessed in secondary endpoints, such as ovarian response, OHSS risk management and gonadotropin consumption.
Ovarian response and total FSH dose: Excessive ovarian response leading to triggering with GnRH agonist occurred for fewer patients with the individualized REKOVELLE dosing regimen compared to the follitropin alfa dosing regimen (p<0.05). Low ovarian response leading to cycle cancellation occurred at comparable rates with REKOVELLE and follitropin alfa.
The overall average number of oocytes retrieved was similar for patients treated with REKOVELLE and follitropin alfa, with more patients treated with REKOVELLE achieving 8-14 oocytes in comparison to follitropin alfa at a starting dose of 11 micrograms (150 IU) and adjustments during stimulation (p<0.05). The average REKOVELLE daily dose was 0.16 μg/kg. The ovarian response and total FSH dose overall and according to AMH concentration are displayed in Table 2. (See Table 2.)
Click on icon to see table/diagram/image
Safety-OHSS risk management: The incidence of patients who required preventive interventions for early OHSS, such as triggering with GnRH agonist or administration of dopamine agonist, was reduced by 50% in the REKOVELLE treated patients compared to the follitropin alfa treated patients (p<0.05). Early OHSS and/or preventive interventions as well as early and late OHSS and/or preventive interventions occurred less frequently with the individualized REKOVELLE dosing regimen compared to the standard follitropin alfa dosing regimen (p<0.05). OHSS risk management parameters are summarized in Table 3. (See Table 3.)
Click on icon to see table/diagram/image
In ovulatory patients with polycystic ovaries, the incidence of early moderate/severe OHSS and/or preventive interventions for early OHSS was 7.7% with REKOVELLE and 26.7% with follitropin alfa.
Safety-immunogenicity: Anti-FSH antibodies were measured pre-dosing and post-dosing in patients undergoing up to three repeated treatment cycles with REKOVELLE (665 patients in cycle 1 in the ESTHER 1 trial as well as 252 patients in cycle 2 and 95 patients in cycle 3 in the ESTHER 2 trial). The incidence of anti-FSH antibodies after treatment with REKOVELLE was 1.1% in cycle 1, 0.8% in cycle 2 and 1.1% in cycle 3. These rates were similar to the incidence of pre-existing anti-FSH antibodies before exposure to REKOVELLE in cycle 1 which was 1.4%, and comparable to the incidences of anti-FSH antibodies after treatment with follitropin alfa. In all patients with anti-FSH antibodies, titres were undetectable or very low and without neutralising capacity.
Repeated treatment with REKOVELLE of patients with pre-existing or treatment-induced anti-FSH antibodies did not increase the antibody titre, was not associated with decreased ovarian response, and did not induce immune-related adverse events.
Pharmacokinetics: The pharmacokinetic profile of follitropin delta has been investigated in healthy female subjects and in IVF/ICSI patients undergoing COS. Following repeated daily subcutaneous administrations, REKOVELLE reaches steady-state within 6 to 7 days with a threefold higher concentration compared with the concentration after the first dose. Circulating levels of follitropin delta are inversely related to the body weight, which supports individualized dosing based on body weight. Follitropin delta leads to greater exposure than follitropin alfa.
Absorption: After daily subcutaneous administration of REKOVELLE, the time to maximum serum concentration is 10 hours. The absolute bioavailability is about 64%.
Distribution: The volume of distribution at steady state is about 9 L. Within the therapeutic dose range, exposure to follitropin delta increases proportionally with the dose.
Elimination: Following intravenous administration, the clearance of follitropin delta is 0.3 L/h. The terminal elimination half-life after single subcutaneous administration is 40 hours and after multiple subcutaneous administration is 28 hours. Comparison of the pharmacokinetics of follitropin delta with follitropin alfa following daily subcutaneous administration of equal doses of IUs for 7 days, revealed that the AUC and Cmax are 1.7-fold and 1.6-fold higher for follitropin delta than for follitropin alfa. Follitropin delta is expected to be eliminated similarly to other follitropins, i.e. mainly by the kidneys. The fraction of follitropin delta excreted unchanged in the urine was estimated to 9%.
Toxicology: Preclinical Safety Data: Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity and local tolerance. The overdose of follitropin delta resulted in pharmacological or exaggerated pharmacological actions. Follitropin delta had a negative effect on fertility and early embryonic development in rats when administered in doses ≥0.8 micrograms/kg/day which is above the recommended maximal dose in humans. The relevance of these findings for the clinical use of REKOVELLE is limited.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image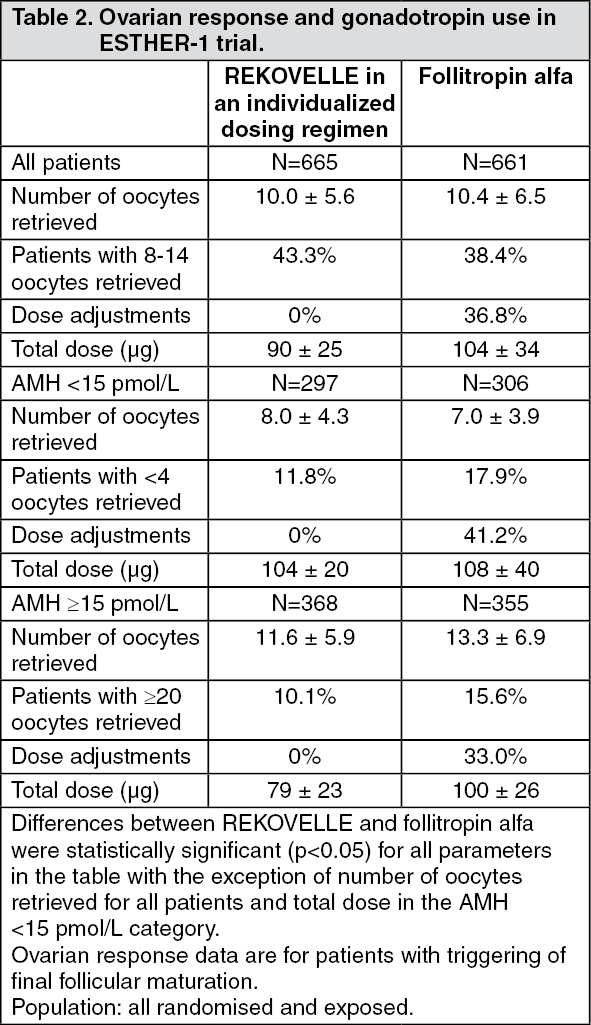 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image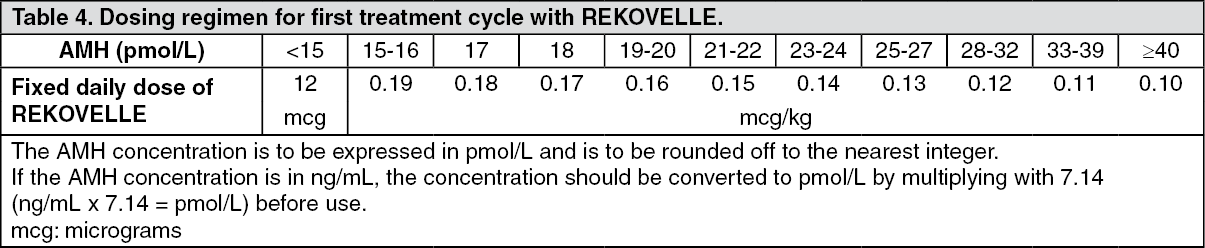 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
