Pharmacotherapeutic group: Immune sera and immunoglobulins: immunoglobulins, normal human, for intravascular administration.
ATC code: J06BA02.
Pharmacology: Pharmacodynamics: Properties/Effects: Privigen is prepared from plasma from 1000 or more human donors. The manufacturing process for Privigen includes the following steps: ethanol precipitation of the IgG plasma fraction, followed by octanoic acid fractionation and incubation at pH 4. Subsequent purification steps comprise depth filtration, chromatography, immunoaffinity chromatography to specifically reduce blood group A and B antibodies (isoagglutinins A and B) and a filtration step that can remove particles to a size of 20 nm.
Privigen contains mainly IgG that are present in the normal human population and that show a broad spectrum of functionally intact antibodies against infectious agents. In the replacement therapy adequate doses of Privigen may restore abnormally low IgG levels to the normal range and thus help against infections.
The IgG subclass distribution in Privigen corresponds roughly to that of native human plasma. Both the Fc and the Fab functions of the IgG molecules are preserved. The ability of the Fab parts to bind antigens was demonstrated with biochemical and biological methods. The Fc function was tested with complement activation and with Fc receptor-mediated leukocyte activation. The inhibition of immune complex-induced complement activation ("scavenging", an anti-inflammatory function of IVIgs) is preserved in Privigen. Privigen does not lead to non-specific activation of the complement system or of prekallikrein.
The mechanism of action in indications other than replacement therapy is not fully elucidated, but includes immunomodulatory effects.
Clinical Eficacy: The safety and efficacy of Privigen was investigated in 7 prospective, open, single-arm, multicentre studies carried out in Europe (ITP, PID and CIDP studies), Japan (PID and CIDP study), and in the US (PID and CIDP study). Further data on safety and efficacy were collected in a prospective, open, single-arm, multicentre extension study with PID patients performed in the US.
PID: In the pivotal study, 80 patients between 3 and 69 years of age with PID were given a Privigen infusion at a median dose of 200-888 mg/kg bw every 3 to 4 weeks for at most 1 year. With this treatment, constant IgG trough levels were achieved over the whole of the treatment period, the mean concentrations being 8.84 g/l to 10.27 g/l. The incidence of acute, severe bacterial infections (aSBI) was 0.08 per patient per year (the upper 97.5% confidence limit was 0.182).
As in the pivotal study, Privigen dosages were administered in the PID extension study to a total of 55 patients (of which 45 had already been treated in the pivotal study and 10 were newly recruited patients). The results of the pivotal study were confirmed for the average IgG trough levels (9.31 g/l to 11.15 g/l) and the rate of aSBI (0.018 per patient per year with an upper 97.5% confidence interval of 0.098).
ITP: 57 patients aged between 15 and 69 years with chronic ITP took part in the ITP study. Their platelet count at the start was 20 x 10
9/l. After administration of Privigen at a dose 1 g/kg bw on two consecutive days, the platelet count rose to at least 50 x 10
9/l within 7 days of the first infusion in 80.7% of the patients. In 43% of the patients, this increase occurred after just one day, before the second infusion. The mean time until this platelet count was reached was 2.5 days. In patients who responded to the treatment, the platelet count remained ≥50 x 10
9/l for a mean period of 15.4 days.
In the second ITP study on patients aged between 18 and 65 years, in 42 subjects (74%) the platelet count increased at least once to ≥50 x 10
9/l within 6 days after the first infusion, which was well within the expected range and similar to response rates were reported for other IVIGs in this indication (70%). A second dose in subjects with platelet counts ≥50 x 10
9/l after the first dose provided a relevant additional benefit in terms of higher and longer-lasting increases in platelet counts compared to a single dose. In subjects with platelet counts <50 x 10
9/l on day 3 receiving a mandatory second infusion, the lowest median platelet count (8.0 x 10
9/l) was observed already at the baseline. In this group, only 30% of subjects were observed with platelet response after the mandatory second dose. Consequently, it was more difficult to increase platelet counts with one infusion in these subjects.
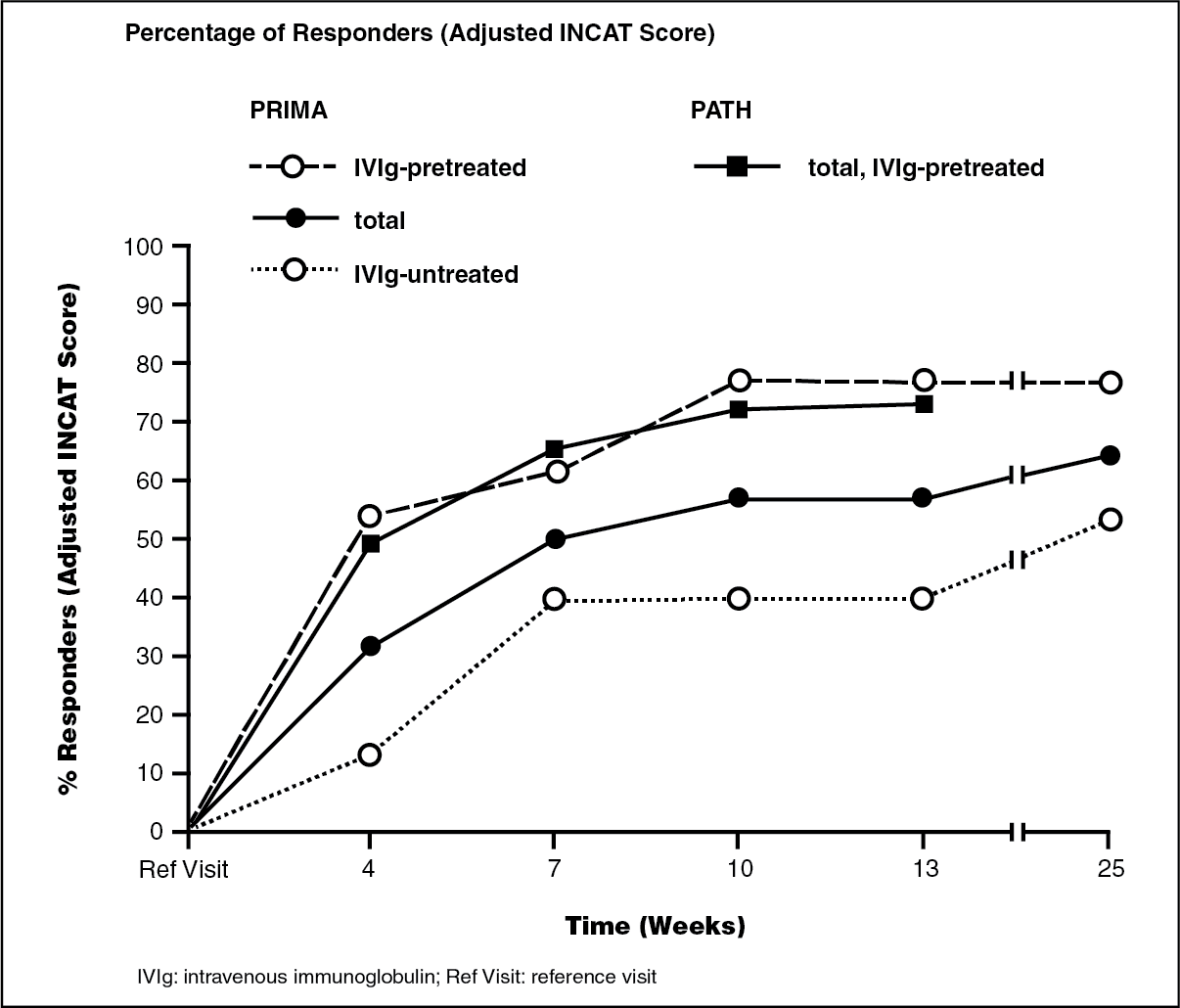
CIDP: In the first CIDP study, a prospective multicenter open label trial PRIMA (Privigen impact on mobility and autonomy study), 28 patients with CIDP (13 subjects with and 15 without IVIg pre-treatment) were treated with a loading dose of 2 g/kg bw given over 2-5 days followed by 6 maintenance doses of 1 g/kg bw given over 1-2 days every 3 weeks. Previously treated patients were withdrawn from IVIg before treatment with Privigen until the deterioration of clinical symptoms was confirmed on the basis of the INCAT scale (Inflammatory Neuropathy Cause and Treatment). On the adjusted 10 point INCAT scale, a clinically meaningful improvement of at least 1-point from baseline to treatment week 25 was observed in 17/28 patients (60.7%, 95% confidence interval 42.41, 76.4). Nine patients responded already after receiving the initial induction dose to the treatment at week 4 and 16 by week 10.
In a second clinical study, a prospective, multicenter, randomized, placebo-controlled PATH [Polyneuropathy and Treatment with Hizentra] study, 207 subjects with CIDP were treated with Privigen in the pre-randomization phase of the study. Subjects all with IVIg pre-treatment of at least 8 weeks and an IVIg-dependence confirmed by clinically evident deterioration during an IVIg withdrawal phase of up to 12 weeks, received a Privigen loading dose of 2 g/kg bw followed by up to 4 Privigen maintenance doses of 1 g/kg bw every 3 weeks for up to 13 weeks.
Following clinical deterioration during IVIg withdrawal, clinical improvement of CIDP was primarily defined by a decrease of ≥1 point at the adjusted INCAT score. Additional measures of CIDP improvement were an R-ODS increase of ≥4 points, a mean grip strength increase of ≥8 kPa, or an MRC sum score increase of ≥3 points. Overall, 91% of subjects (188 patients) showed improvement in at least one of the criteria above by week 13.
By adjusted INCAT score, the responder rate by week 13 was 72.9% (151/207 patients), with 149 patients responding already by week 10. A total of 43 of the 207 patients achieved a better CIDP status as assessed by the adjusted INCAT score compared to their CIDP status at study entry.
The comparability of the response rates and mean adjusted INCAT scores for the IVIg pretreated subjects in both PRIMA and PATH study are shown in the figure as follows.
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The mean improvement at the end of the treatment period compared to reference visit was 1.4 points in the PRIMA (1.8 points in IVIg pretreated subjects) and 1.2 points in PATH study.
In PRIMA, the percentage of responders in the overall Medical Research Council (MRC) score (defined as an increase by ≥3 points) was 85% (87% in the IVIg-untreated and 82% in IVIg-pretreated) and 57% in PATH. The overall median time to first MRC sum score response in PRIMA was 6 weeks (6 weeks in the IVIg-untreated and 3 weeks in the IVIg-pretreated) and 9.3 weeks in PATH. MRC sum score in PRIMA improved by 6.9 points (7.7 points for IVIg-untreated and 6.1 points for IVIg-pretreated) and by 3.6 points in PATH.
The grip strength of the dominant hand improved by 14.1 kPa (17.0 kPa in IVIg-untreated and 10.8 kPa in IVIg-pretreated subjects) in the PRIMA study, while in PATH the grip strength of the dominant hand improved by 12.2 kPa. For the non-dominant hand similar results were observed in both studies, PRIMA and PATH.
The efficacy and safety profile in the PRIMA and the PATH study in CIDP patients were overall comparable.
Paediatric population: No differences were observed in the pharmacodynamic properties and safety profile between adult and paediatric study patients.
Pharmacokinetics: Privigen is immediately and completely bioavailable in the recipient's circulation after intravenous administration. It is distributed relatively quickly between plasma and extravascular fluid. Equilibrium between the intravascular and extravascular compartments is reached after approximately 3 to 5 days.
IgG and IgG complexes are broken down in the cells of the reticuloendothelial system. The half-life may vary from patient to patient.
The pharmacokinetic parameters for Privigen were determined in both clinical studies in patients with primary immunodeficiency syndrome (see Properties/Effects as previously mentioned). 25 patients (aged 13 to 69 years) in the pivotal study and 13 patients (aged 9 to 59 years) in an extension of this study participated in the pharmacokinetic (PK) assessment (see Table 1 as follows).
Click on icon to see table/diagram/image
In the pivotal study, the median half-life of Privigen in primary immunodeficiency patients was 36.6 days and 31.1 days in the extension of this study.
Paediatric population: No differences were seen in the pharmacokinetic parameters between adult and paediatric study patients with PID. There are no data on pharmacokinetic properties in paediatric patients with CIDP.
Toxicology: Preclinical data: The safety of Privigen has been investigated in several preclinical studies with particular reference to the excipient L-proline. L-proline is a physiological, non-essential amino acid.
Studies in rats given daily L-proline doses of 1450 mg/kg bw did not show any evidence of teratogenicity or embryotoxicity. Genotoxicity studies of L-proline did not show any pathological findings.
Some published studies pertaining to hyperprolinaemia have shown that long-term, high doses of L-proline have effects on brain development in very young rats. However, in studies where the dosing was designed to reflect the clinical indications for Privigen, no effects on brain development were observed. Further safety-pharmacology studies of L-proline in adult and juvenile rats did not reveal behavioural disorders.
Immunoglobulins are natural components of the human body. Data from animal testing of acute and chronic toxicity and embryofoetal toxicity of immunoglobulins are inconclusive on account of interactions between immunoglobulins from heterogeneous species and the induction of antibodies to heterologous proteins. In local tolerability studies in rabbits in which Privigen was administered intravenously, paravenously, intra-arterially, and subcutaneously, the product was well tolerated.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image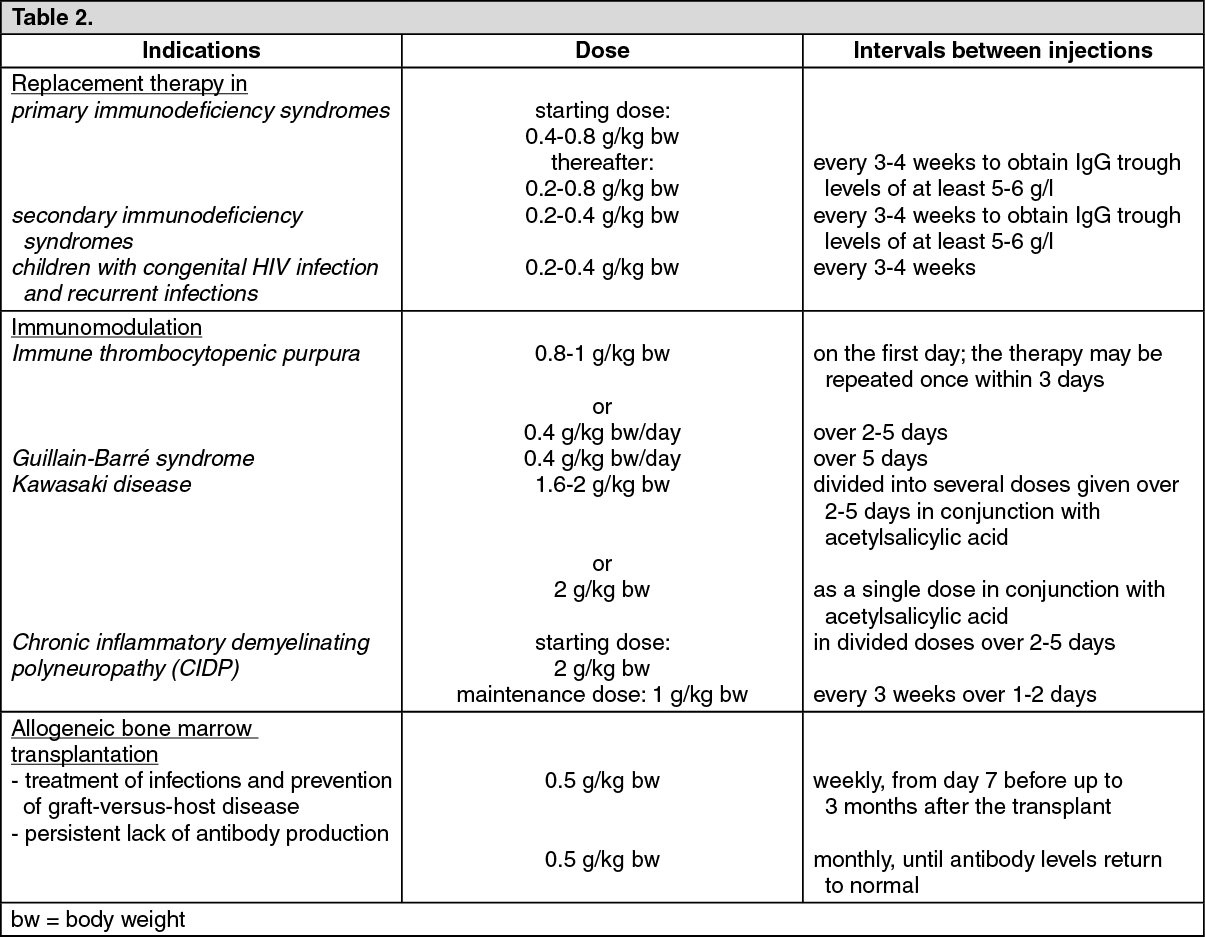 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image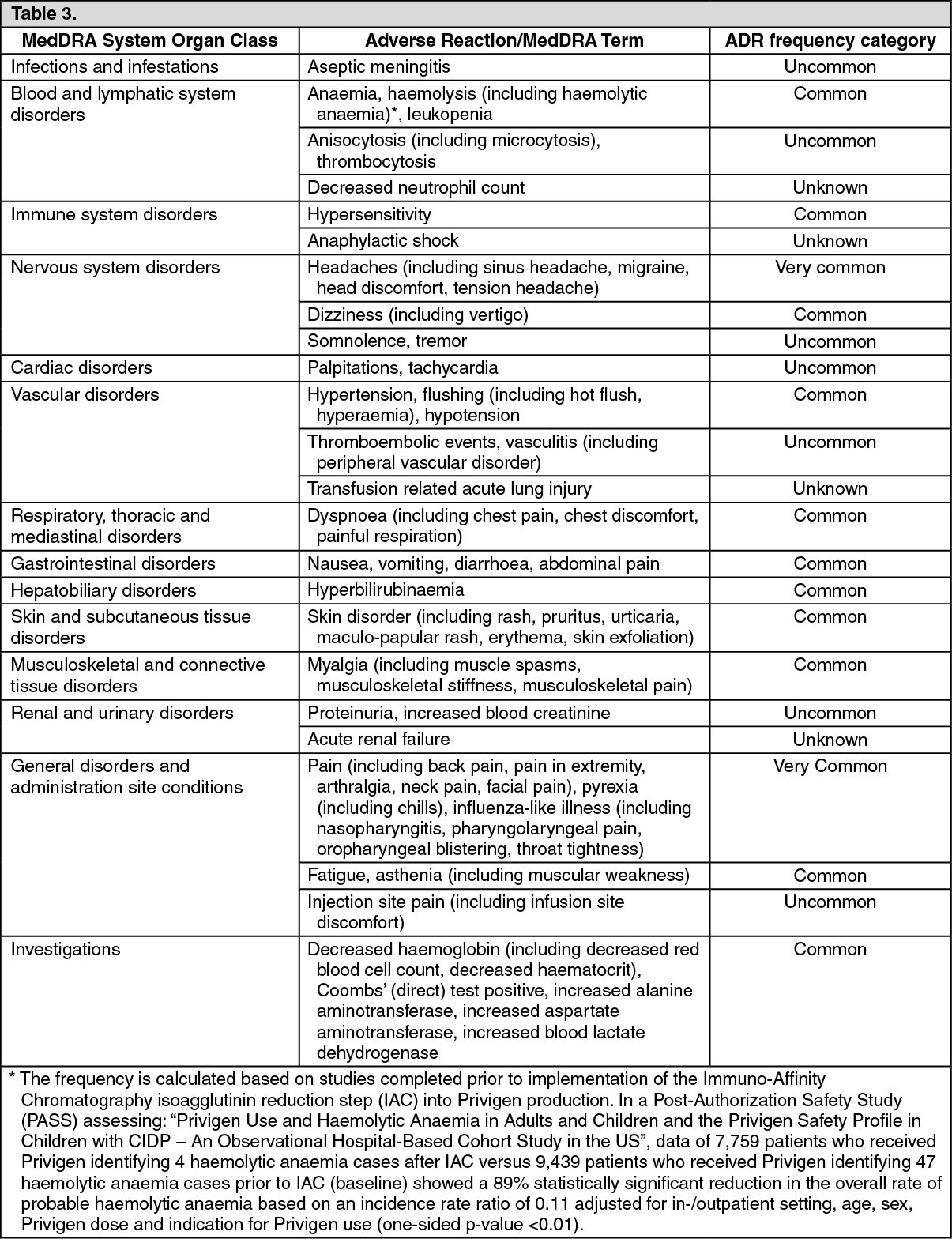 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
