


 Sign Out
Sign Out


Pharmacology: MOBIC is a non-steroidal anti-inflammatory drug (NSAID) of the enolic acid class, which has shown anti-inflammatory, analgesic and antipyretic properties in animals. Meloxicam showed potent anti-inflammatory activity in all standard models of inflammation. A common mechanism for the effects as follows may exist in the ability of meloxicam to inhibit the biosynthesis of prostaglandins, known mediators of inflammation.
Comparison of the ulcerogenic dose and the anti-inflammatory effective dose in the rat adjuvant arthritis model confirmed a superior therapeutic margin in animals over standard NSAIDs. In vivo, meloxicam inhibited prostaglandin biosynthesis more potently at the site of inflammation than in the gastric mucosa or the kidney.
These differences are thought to be related to a selective inhibition of COX-2 relative to COX-1 and it is believed that COX-2 inhibition provides the therapeutic effects of NSAIDs whereas inhibition of constitutive COX-1 may be responsible for gastric and renal side effects.
The COX-2 selectivity of meloxicam has been confirmed both in vitro and ex vivo in a number of test systems. In the human whole blood assay, meloxicam has been shown in vitro to inhibit COX-2 selectively. Meloxicam (7.5 and 15 mg) demonstrated a greater inhibition of COX-2 ex vivo, as demonstrated by a greater inhibition of lipopolysaccharide-stimulated PGE2 production (COX-2) as compared with thromboxane production in clotting blood (COX-1). These effects were dose-dependent. Meloxicam has been demonstrated to have no effect on either platelet aggregation or bleeding time at recommended doses ex vivo, while indomethacin, diclofenac, ibuprofen and naproxen significantly inhibited platelet aggregation and prolonged bleeding.
Clinical Trials: In clinical trials, gastro-intestinal adverse events overall were reported less frequently with meloxicam 7.5 mg and 15 mg than with the NSAIDs with which it has been compared, due predominantly to a lower reporting incidence of events such as dyspepsia, vomiting, nausea and abdominal pain. The incidence of upper gastro-intestinal perforation, ulcers, and bleeds reported in association with meloxicam is low and dose dependent.
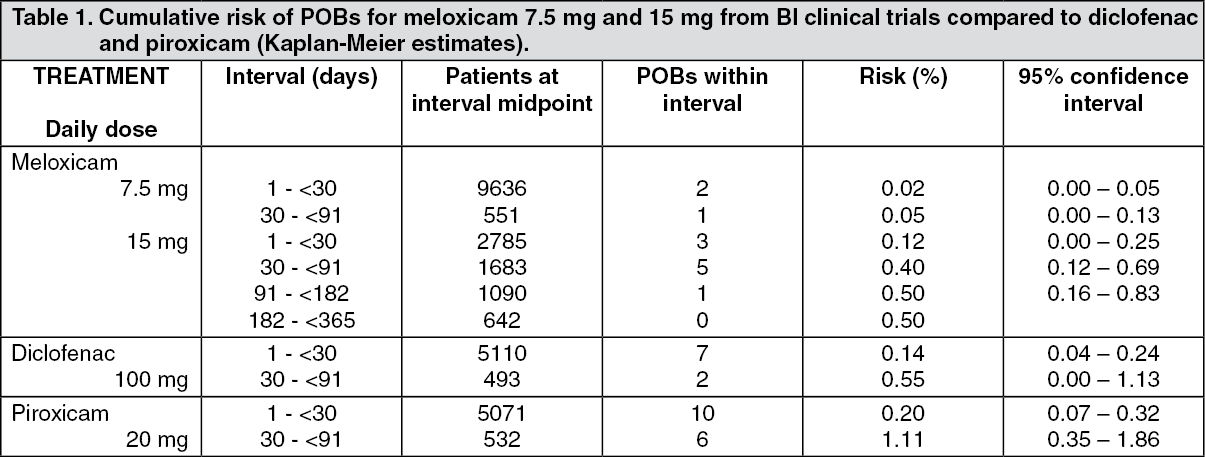
There is no single study powered adequately to detect statistically differences in the incidence of clinically significant upper gastro-intestinal perforation, obstruction, or bleeds between meloxicam and other NSAIDs. A pooled analysis has been conducted involving patients treated with meloxicam in 35 clinical trials in the indications osteoarthritis, rheumatoid arthritis, and ankylosing spondylitis. Exposure to meloxicam in these trials ranged from 3 weeks to one year (most patients were enrolled in one-month studies). Almost all patients participated in trials that permitted enrolment of patients with a prior history of gastro-intestinal perforation, ulcer or bleed.
The incidence of clinically significant upper gastro-intestinal perforation, obstruction, or bleed (POB) was assessed retrospectively following independent blinded review of cases. Results are shown in the following table. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePharmacokinetics: Absorption: Meloxicam is well absorbed from the gastro-intestinal tract, which is reflected by a high absolute bioavailability of about 90% following oral administration.
Following single dose administration of meloxicam, median maximum plasma concentrations are achieved within 2 hours for the suspension and within 5-6 hours with solid oral dosage forms (tablets).
Extent of absorption for meloxicam following oral administration is not altered by concomitant food intake or the use of inorganic antacids. Dose linearity was demonstrated after oral administration in the therapeutic dose range of 7.5 to 15 mg.
With multiple dosing, steady state conditions were reached within 3 to 5 days.
Once daily dosing leads to mean drug plasma concentrations with a relatively small peak-trough fluctuation in the range of 0.4 - 1.0 μg/mL for 7.5 mg doses and 0.8 - 2.0 μg/mL for 15 mg doses, respectively (Cmin and Cmax at steady state, correspondingly).
Mean maximum plasma concentrations of meloxicam at steady state, are achieved within five to six hours for the tablet. Extent of absorption for meloxicam following oral administration is not altered by concomitant food intake.
Distribution: Meloxicam is very strongly bound to plasma proteins, essentially albumin (99%).
Meloxicam penetrates into synovial fluid to give concentrations approximately half of those in plasma.
Volume of distribution is low, i.e. approx. 11 L after i.m. or i.v. administration, and shows interindividual variation in the order of 7 - 20%.
The volume of distribution following administration of multiple oral doses of meloxicam (7.5 to 15 mg) is about 16 L with coefficients of variation ranging from 11 to 32%.
Biotransformation: Meloxicam undergoes extensive hepatic biotransformation.
Four different metabolites of meloxicam were identified in urine, which are all pharmacodynamically inactive.
The major metabolite, 5'-carboxymeloxicam (60% of dose), is formed by oxidation of an intermediate metabolite 5'-hydroxymethylmeloxicam, which is also excreted to a lesser extent (9% of dose). In vitro studies suggest that CYP 2C9 plays an important role in this metabolic pathway, with a minor contribution from the CYP 3A4 isoenzyme. The patient's peroxidase activity is probably responsible for the other two metabolites, which account for 16% and 4% of the administered dose respectively.
Elimination: Meloxicam is excreted predominantly in the form of metabolites and occurs to equal extents in urine and faeces. Less than 5% of the daily dose is excreted unchanged in faeces, while only traces of the parent compound are excreted in urine.
The mean elimination half-life varies between 13 and 25 hours after oral, i.m. and i.v. administration.
Total plasma clearance amounts about 7-12 mL/min following single doses orally, intravenously or rectally administered.
Linearity/non-linearity: Meloxicam demonstrates linear pharmacokinetics in the therapeutic dose range of 7.5 mg to 15 mg following per oral administration.
Special populations: Patients with hepatic/renal insufficiency: Neither hepatic insufficiency, nor mild renal insufficiency has a substantial effect on meloxicam pharmacokinetics. Subjects with moderate renal impairment had significantly higher total drug clearance. A reduced protein binding is observed in patients with terminal renal failure. In terminal renal failure, the increase in the volume of distribution may result in higher free meloxicam concentrations.
Elderly: Elderly male subjects exhibited similar mean pharmacokinetic parameters compared to those of young male subjects. Elderly female patients showed higher AUC-values and longer elimination half-lives compared to those of young subjects of both genders. Mean plasma clearance at steady state in elderly subjects was slightly lower than that reported for younger subjects.