Pharmacotherapeutic group: Antithrombotic agents, enzymes.
ATC code: B01AD11.
Pharmacology: Mode of action: Tenecteplase is a recombinant fibrin-specific plasminogen activator that is derived from native t-PA by modifications at three sites of the protein structure. It binds to the fibrin component of the thrombus (blood clot) and selectively converts thrombus-bound plasminogen to plasmin, which degrades the fibrin matrix of the thrombus. Tenecteplase has higher fibrin specificity and greater resistance to inactivation by its endogenous inhibitor (PAI-1) compared to native t-PA.
Pharmacodynamics: After administration of tenecteplase dose dependent consumption of α2-antiplasmin (the fluid-phase inhibitor of plasmin) with consequent increase in the level of systemic plasmin generation have been observed. This observation is consistent with the intended effect of plasminogen activation. In comparative studies a less than 15% reduction in fibrinogen and a less than 25% reduction in plasminogen were observed in subjects treated with the maximum dose of tenecteplase (10,000 U, corresponding to 50 mg), whereas alteplase caused an approximately 50% decrease in fibrinogen and plasminogen levels. No clinically relevant antibody formation was detected at 30 days.
Clinical trials: Patency data from the phase I and II angiographic studies suggest that tenecteplase, administered as a single intravenous bolus, is effective in dissolving blood clots in the infarct-related artery of subjects experiencing an AMI on a dose related basis.
ASSENT 2 Study: A large scale mortality trial (ASSENT 2) in approx. 17,000 patients showed that tenecteplase is therapeutically equivalent to alteplase in reducing mortality (6.2% for both treatments, at 30 days) and that the use of tenecteplase is associated with a significantly lower incidence of non-intracranial bleedings (26.4% vs. 28.9%, p=0.0003). The reduction of the risk of bleeding is likely to be related to the increased fibrin specificity of tenecteplase and to its weight adapted regimen.
This translates into a significantly lower need of transfusions (4.3% vs. 5.5%, p=0.0002). Intracranial haemorrhage occurred at a rate of 0.93% vs. 0.94% for tenecteplase and alteplase, respectively. In the 475 patients treated beyond 6 hours numerical differences in favour of tenecteplase were observed with regard to 30-day mortality (4.3% vs. 9.6%), stroke (0.4% vs. 3.3%) and ICH (0% vs. 1.7%).
ASSENT 3 Study: The ASSENT 3 study aimed to optimise tenecteplase concomitant antithrombotic therapy towards both improving early patency rates and maintaining perfusion, mainly to overcome the paradoxical pro-coagulant effect arising from the release by clot lysis of trapped thrombin. Three different concomitant antithrombotic regimens were compared in 6,095 patients: Full-dose tenecteplase + unfractionated heparin (UFH) versus full-dose tenecteplase + low molecular weight (LMW) heparin (enoxaparin) versus half-dose tenecteplase + unfractionated heparin + full dose abciximab. UFH was used as recommended by AHA/ACC guidelines according to a full body-weight adapted low dose regimen as follows: A single i.v. bolus of 60 IU/kg (maximum 4000 IU) immediately followed by an intravenous infusion of 12 IU/kg/hr (maximum 1000 IU/hr) for the first 3 hours, thereafter according to aPTT monitoring for up to 48 hours to maintain aPTT at 50-70 seconds.
30-day mortality rates are respectively 6.0%, 5.4% and 6.6%, the in-hospital major bleeds (other than ICH) 2.16%, 3.04% and 4.32%.and the intracranial haemorrhage 0.93%, 0.88% and 0.94%.
The recommended ACC/AHA fully body-weight adjusted low dose unfractionated heparin regimen used in ASSENT 3 concomitantly with tenecteplase results in less systemic bleeding but similar ICH rates compared to the more aggressive unfractionated heparin regimen dosing used in ASSENT 2 without loss of efficacy.
ASSENT 3 PLUS study: ASSENT 3 PLUS, a satellite study of ASSENT 3, was designed to investigate the pre-hospital setting. The efficacy and safety of full-dose tenecteplase + unfractionated heparin versus full-dose tenecteplase + low molecular weight (LMW) heparin (enoxaparin) has been evaluated in 1639 patients.
The study design and treatments dosage used are identical to those of the ASSENT 3 study. Pre-hospital reperfusion therapy with tenecteplase and UFH or enoxaparin allowed treatment within 2 hours of symptom onset in >50% of the patients with STEMI.
From ASSENT 3 and 3 PLUS studies, pre-hospital as well as in-hospital adjunctive therapy with enoxaparin reduced the incidence of ischemic complications as compared to adjunctive therapy with UFH: the incidence of 30-day efficacy composite endpoint (death, re-infarction, refractory ischaemia) was respectively 11.4% versus 15.4% in ASSENT 3 and 14.2% versus 17.4% in ASSENT 3 PLUS. However, in the pre-hospital setting, tenecteplase with enoxaparin at the dose used was associated with an increased risk of major bleeding and ICH in patients >75 years of age.
Coronary patency and limited clinical outcome data showed that AMI patients have been successfully treated later than 6 hours after symptom onset.
ASSENT-4 PCI study: The ASSENT-4 PCI study was designed to show if in 4000 patients with large myocardial infarctions pre-treatment with full dose tenecteplase and concomitant single bolus of up to 4,000 IU unfractionated heparin administered prior to primary Percutaneous Coronary Intervention (PCI) to be performed within 60 to 180 minutes leads to better outcomes than primary PCI alone. The trial was prematurely terminated with 1667 randomised patients due to a numerically higher mortality in the facilitated PCI group receiving tenecteplase. The occurrence of the primary endpoint, a composite of death or cardiogenic shock or congestive heart failure within 90 days, was significantly higher in the group receiving the exploratory regimen of tenecteplase followed by routine immediate PCI: 18.6% (151/810) compared to 13.4% (110/819) in the PCI only group, p=0.0045. This significant difference between the groups for the primary endpoint at 90 days was already present in-hospital and at 30 days. Numerically, all of the components of the clinical composite endpoint were in favour of the PCI only regimen: death: 6.7% versus 4.9% p=0.14; cardiogenic shock: 6.3% versus 4.8% p=0.19; congestive heart failure: 12.0% versus 9.2% p=0.06 respectively. The secondary endpoints re-infarction and repeat target vessel revascularisation were significantly increased in the group pre-treated with tenecteplase: reinfarction: 6.1% versus 3.7% p=0.0279; repeat target vessel revascularisation: 6.6% versus 3.4% p=0.0041. The following adverse events occurred more frequently with tenecteplase prior to PCI: intracranial haemorrhage: 1% versus 0% p=0.0037; stroke: 1.8% versus 0% p<0.0001; major bleeds: 5.6% versus 4.4% p=0.3118; minor bleeds: 25.3% versus 19.0% p=0.0021; blood transfusions: 6.2% versus 4.2% p=0.0873; abrupt vessel closure: 1.9% versus 0.1% p=0.0001.
STREAM study:
The STREAM study was designed to evaluate the efficacy and safety of a pharmaco-invasive strategy of early fibrinolytic treatment with tenecteplase and additional antiplatelet and anticoagulant therapy followed by angiography within 6-24 hours or rescue coronary intervention versus a strategy of standard primary PCI.
The study population consisted of patients with ST elevation acute myocardial infarction within 3 hours of onset of symptoms not able to undergo primary PCI within one hour of first medical contact.
A sample size of approximately 1000 patients per treatment group was planned for this exploratory study. After 382 patients had been enrolled (19.5 % of the planned study population), the dose of the tenecteplase bolus was reduced by half for the patients ≥75 years because of a higher incidence of intracranial haemorrhage (ICH) in this sub-group.
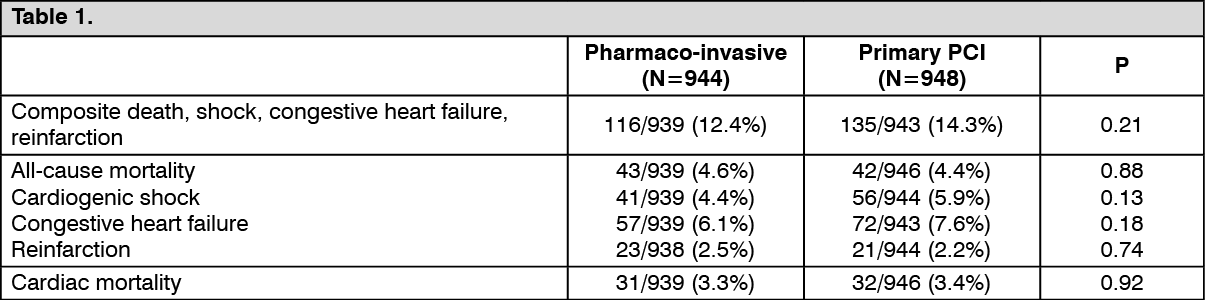
1892 patients were randomised by means of an interactive voice response system. The primary endpoint, a composite of death or cardiogenic shock or congestive heart failure or re-infarction within 30 days was observed in 12.4% (116/939) of the pharmaco-invasive arm versus 14.3% (135/943) in the primary PCI arm [relative risk 0.86 (0.68-1.09)].
Single components of the primary composite endpoint for the pharmaco-invasive strategy versus primary PCI respectively were observed with the following frequencies (see Table 1):
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The observed incidence of major and of minor non-ICH bleeds were similar in both groups (see Table 2):
Click on icon to see table/diagram/image
Incidence of total strokes and intracranial haemorrhage (see Table 3):
Click on icon to see table/diagram/image
None of the differences between groups displayed in the above tables reached the threshold of statistical significance except for the incidence of total strokes and ICH, however the incidences in the pharmaco-invasive group were as observed in previous clinical studies.
After the dose reduction of tenecteplase by half in patients ≥75 years there was no further intracranial hemorrhage (0 of 97 patients) (95% CI: 0.0-3.7) versus 8.1% (3 of 37 patients) (95% CI: 1.7-21.9) prior to the dose reduction. The bounds of the confidence interval of the observed incidences prior and after dose reduction are overlapping.
In patients ≥75 years the observed incidence of the primary efficacy composite end point for the pharmaco-invasive strategy and primary PCI were as follows: before dose reduction 11/37 (29.7%) (95% CI: 15.9-47.0) vs. 10/32 (31.3%) (95% CI: 16.1-50.0), after dose reduction: 25/97 (25.8%) (95% CI: 17.4-35.7) vs. 25/88 (24.8%) (95% CI: 19.3-39.0). In both groups the bounds of the confidence interval of the observed incidences prior and post dose reduction are overlapping.
Pharmacokinetics: Absorption and Distribution: Tenecteplase is an intravenously administered, recombinant protein that activates plasminogen. Following i.v. bolus administration of 30 mg tenecteplase in patients with acute myocardial infarction, the initially estimated tenecteplase plasma concentration was 6.45 ± 3.60 μg/mL (mean ±SD). The distribution phase represents 31% ± 22% to 69% ± 15% (mean ±SD) of the total AUC following the administration of doses ranges from 5 to 50 mg.
Data on tissue distribution were obtained in studies with radioactively labelled tenecteplase in rats. The main organ to which tenecteplase distributed was the liver. It is not known whether and to which extent tenecteplase binds to plasma proteins in humans. The mean residence time (MRT) in the body is approximately 1 h and the mean (±SD) volume of distribution at the steady-state (Vss) ranged from 6.3 ± 2 L to 15 ± 7 L.
Metabolism: Tenecteplase is cleared from the circulation by binding to specific receptors in the liver followed by catabolism to small peptides. Binding to hepatic receptors is, however, reduced compared to native t-PA, resulting in a prolonged half-life.
After single intravenous bolus injection of tenecteplase in patients with acute myocardial infarction, tenecteplase antigen exhibits biphasic elimination from plasma. There is no dose dependence of tenecteplase clearance in the therapeutic dose range. The initial, dominant half-life is 24 ± 5.5 (mean ± SD) min, which is 5 times longer than native t-PA.
The terminal half-life is 129 ± 87 min, and plasma clearance is 119 ± 49 ml/min.
Increasing body weight resulted in a moderate increase of tenecteplase clearance, and increasing age resulted in a slight decrease of clearance. Women exhibit in general lower clearance than men, but this can be explained by the generally lower body weight of women.
Linearity/Non-Linearity: The dose linearity analysis based on AUC suggested that tenecteplase exhibits non-linear pharmacokinetics in the dose range studied, i.e. 5 to 50 mg.
Special Populations: Renal and hepatic impairment: Because elimination of tenecteplase is through the liver, it is not expected that renal dysfunction will affect the pharmacokinetics of METALYSE. This is also supported by animal data. However, the effect of renal and hepatic dysfunction on pharmacokinetics of tenecteplase in humans has not been specifically investigated.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image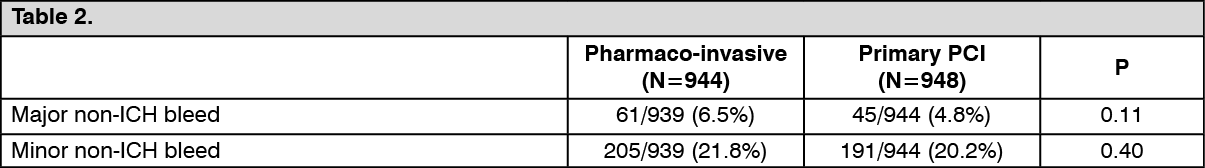 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image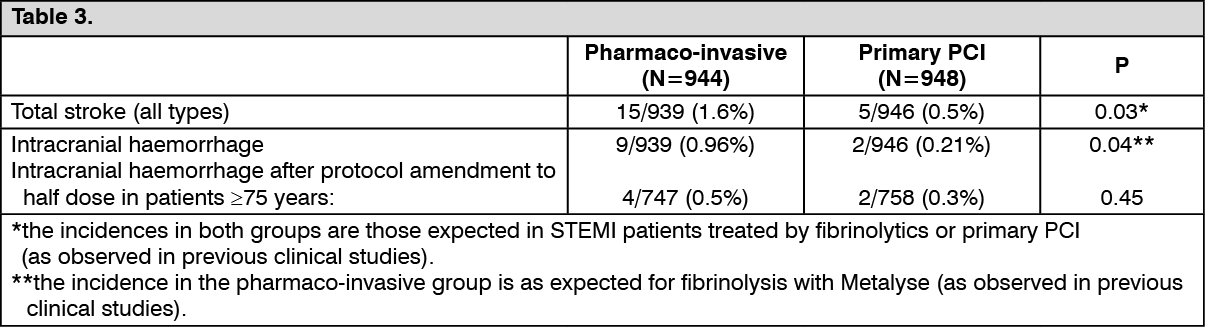 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image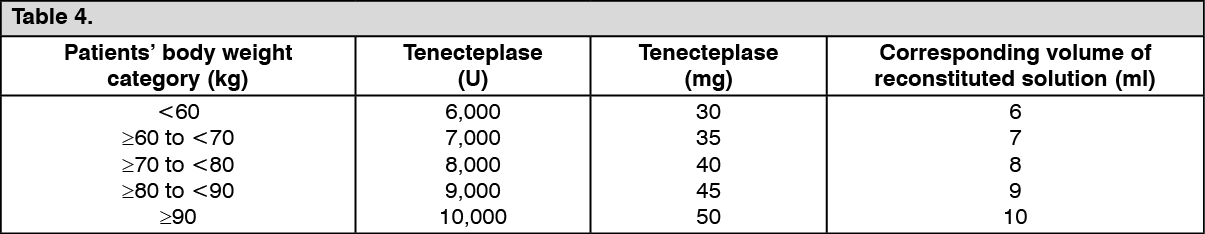 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image 50 mg (10_000 U)c2a7437a-4a4c-46de-8923-9faa0009bc74.GIF)

 Sign Out
Sign Out
