Pharmacotherapeutic group: Selective Immunosuppressants.
ATC code: L04AA40.
Pharmacology: Pharmacodynamics: Mechanism of action: Cladribine is a nucleoside analogue of deoxyadenosine. A chlorine substitution in the purine ring protects cladribine from degradation by adenosine deaminase, increasing the intracellular residence time of the cladribine prodrug. Subsequent phosphorylation of cladribine to its active triphosphate form, 2-chlorodeoxyadenosine triphosphate (Cd-ATP), is particularly efficiently achieved in lymphocytes, due to their constitutively high deoxycytidine kinase (DCK) and relatively low 5'-nucleotidase (5'-NTase) levels. A high DCK to 5'-NTase ratio favours the accumulation of Cd-ATP, making lymphocytes particularly susceptible to cell death. As a result of a lower DCK/5'-NTase ratio other bone marrow derived cells are less affected than lymphocytes. DCK is the rate limiting enzyme for conversion of the cladribine prodrug into its active triphosphate form, leading to selective depletion of dividing and non-dividing T and B cells.
The primary apoptosis-inducing mechanism of action of Cd-ATP has direct and indirect actions on DNA synthesis and mitochondrial function. In dividing cells, Cd-ATP interferes with DNA synthesis via inhibition of ribonucleotide reductase and competes with deoxyadenosine triphosphate for incorporation into DNA by DNA polymerases. In resting cells cladribine causes DNA single-strand breaks, rapid nicotinamide adenine dinucleotide consumption, ATP depletion and cell death. There is evidence that cladribine can also cause direct caspase-dependent and -independent apoptosis via the release of cytochrome c and apoptosis-inducing factor into the cytosol of non-dividing cells.
MS pathology involves a complex chain of events in which different immune cell types, including autoreactive T and B cells play a key role. The mechanism by which cladribine exerts its therapeutic effects in MS is not fully elucidated but its predominant effect on B and T lymphocytes is thought to interrupt the cascade of immune events central to MS.
Variations in the expression levels of DCK and 5'-NTases between immune cell subtypes may explain differences in immune cell sensitivity to cladribine. Because of these expression levels, cells of the innate immune system are less affected than cells of the adaptive immune system.
Pharmacodynamic effects: Cladribine has been shown to exert long-lasting effects by preferentially targeting lymphocytes and the autoimmune processes involved in the pathophysiology of MS.
Across studies, the largest proportion of patients with grade 3 or 4 lymphopenia (<500 to 200 cells/mm
3 or <200 cells/mm
3) was seen 2 months after the first cladribine dose in each year, indicating a time gap between cladribine plasma concentrations and the maximum haematological effect.
Across clinical studies, data with the proposed cumulative dose of 3.5 mg/kg body weight show a gradual improvement in the median lymphocyte counts back to the normal range at week 84 from the first dose of cladribine (approximately 30 weeks after the last dose of cladribine). The lymphocyte counts of more than 75% of patients returned to the normal range by week 144 from the first dose of cladribine (approximately 90 weeks after the last dose of cladribine).
Treatment with oral cladribine leads to rapid reductions in circulating CD4+ and CD8+ T cells. CD8+ T cells have a less pronounced decrease and a faster recovery than CD4+ T cells, resulting in a temporarily decreased CD4 to CD8 ratio. Cladribine reduces CD19+ B cells and CD16+/CD56+ natural killer cells, which also recover faster than CD4+ T cells.
Clinical efficacy and safety: Relapsing-remitting MS: Efficacy and safety of oral cladribine were evaluated in a randomised, double-blind, placebo-controlled clinical study (CLARITY) in 1,326 patients with relapsing-remitting MS. Study objectives were to evaluate the efficacy of cladribine versus placebo in reducing the annualised relapse rate (ARR) (primary endpoint), slowing disability progression and decreasing active lesions as measured by MRI.
Patients received either placebo (n = 437), or a cumulative dose of cladribine of 3.5 mg/kg (n = 433) or 5.25 mg/kg body weight (n = 456) over the 96-week (2-year) study period in 2 treatment courses. Patients randomised to the 3.5 mg/kg cumulative dose received a first treatment course at weeks 1 and 5 of the first year and a second treatment course at weeks 1 and 5 of the second year. Patients randomised to the 5.25 mg/kg cumulative dose received additional treatment at weeks 9 and 13 of the first year. The majority of patients in the placebo (87.0%) and the cladribine 3.5 mg/kg (91.9%) and 5.25 mg/kg (89.0%) treatment groups completed the full 96 weeks of the study.
Patients were required to have at least 1 relapse in the previous 12 months. In the overall study population, the median age was 39 years (range 18 to 65), and the female to male ratio was approximately 2:1. The mean duration of MS prior to study enrolment was 8.7 years, and the median baseline neurological disability based on Kurtzke Expanded Disability Status Scale (EDSS) score across all treatment groups was 3.0 (range 0 to 6.0). Over two thirds of the study patients were treatment-naive for MS disease-modifying drugs (DMDs). The remaining patients were pre-treated with either interferon beta-1a, interferon beta-1b, glatiramer acetate or natalizumab.
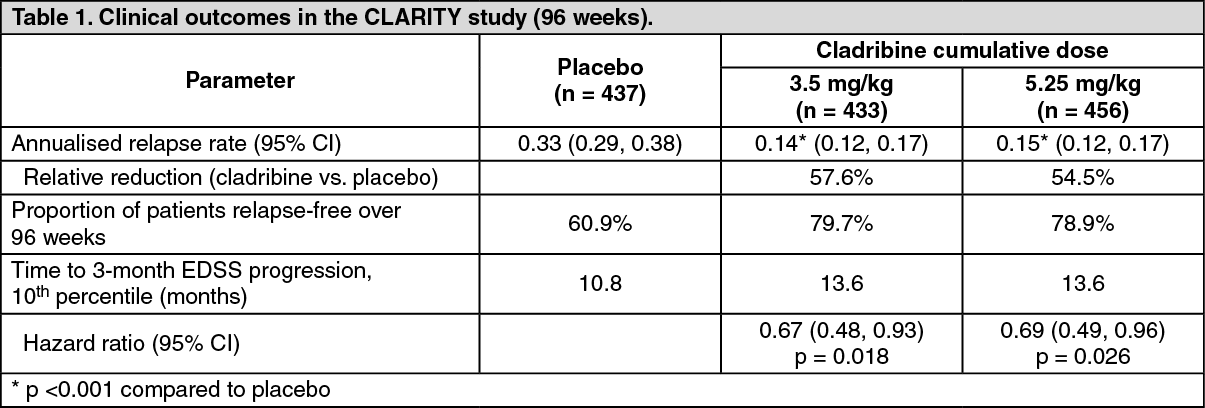
Patients with relapsing-remitting MS receiving cladribine 3.5 mg/kg showed statistically significantly improvements in the annualised relapse rate, proportion of patients relapse-free over 96 weeks, proportion of patients free of sustained disability over 96 weeks and time to 3-month EDSS progression compared to patients on placebo (see Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In addition, the cladribine 3.5 mg/kg treatment group was statistically significantly superior to placebo with regard to number and relative reduction of T1 Gd+ lesions, active T2 lesions and combined unique lesions as demonstrated in brain MRI over the entire 96 weeks of the study. Patients taking cladribine compared to the placebo treatment group had 86% relative reduction in the mean number of T1 Gd+ lesions (adjusted mean number for cladribine 3.5 mg/kg, and placebo groups were 0.12 and 0.91, respectively), 73% relative reduction in the mean number of active T2 lesions (adjusted mean number for cladribine 3.5 mg/kg, and placebo groups were 0.38 and 1.43, respectively) and 74% relative reduction in the mean number of combined unique lesions per patient per scan (adjusted mean number for cladribine 3.5 mg/kg, and placebo groups were 0.43 and 1.72, respectively) (p <0.001 across all 3 MRI outcomes).
Post-hoc analysis of time to 6-month confirmed EDSS progression resulted in a 47% reduction of the risk of disability progression in the cladribine 3.5 mg/kg compared to placebo (hazard ratio = 0.53, 95% CI [0.36, 0.79], p <0.05); in the placebo group the 10
th percentile was reached at 245 days, and not reached at all during the study period in the cladribine 3.5 mg/kg group.
As shown previously in Table 1, higher cumulative doses did not add any clinically meaningful benefit, but were associated with a higher incidence in ≥grade 3 lymphopenia (44.9% in the 5.25 mg/kg group vs. 25.6% in the 3.5 mg/kg group).
Patients who had completed the CLARITY study could be enrolled in CLARITY Extension. In this extension study, 806 patients received either placebo or a cumulative dose of cladribine 3.5 mg/kg (in a regimen similar to that used in CLARITY) over the 96-week study period. The primary objective of this study was safety, while efficacy endpoints were exploratory.
The magnitude of the effect in reducing the frequency of relapses and slowing disability progression in patients receiving the 3.5 mg/kg dose over 2 years was maintained in years 3 and 4 (see Dosage & Administration).
Efficacy in patients with high disease activity: Post-hoc subgroup efficacy analyses have been conducted in patients with high disease activity treated with oral cladribine at the recommended 3.5 mg/kg cumulative dose. These included: patients with 1 relapse in the previous year and at least 1 T1 Gd+ lesion or 9 or more T2 lesions, while on therapy with other DMDs; patients with 2 or more relapses in the previous year, whether on DMD treatment or not.
In the analyses of the CLARITY data, a consistent treatment effect on relapses was observed with the annualised relapse rate ranging from 0.16 to 0.18 in the cladribine groups and 0.47 to 0.50 in the placebo group (p <0.0001). Compared to the overall population, a greater effect was observed in time to 6-month sustained disability where cladribine reduced the risk of disability progression by 82% (hazard ratio = 0.18, 95% CI [0.07, 0.47]). For placebo, the 10
th percentile for disability progression was reached between 16 and 23 weeks, while for the cladribine groups it was not reached during the entire study.
Secondary progressive MS with relapses: A supportive study in patients treated with cladribine as an add-on to interferon beta vs. placebo + interferon beta also included a limited number of patients with secondary progressive MS (26 patients). In these patients, treatment with cladribine 3.5 mg/kg resulted in a reduction of the annualised relapse rate compared to placebo (0.03 versus 0.30, risk ratio: 0.11, p <0.05). There was no difference in annualised relapse rate between patients with relapsing-remitting MS and patients with secondary progressive MS with relapses. An effect on disability progression could not be shown in either subgroup.
Patients with secondary progressive MS were excluded in the CLARITY study. However, a post-hoc analysis of a mixed cohort including CLARITY and ONWARD patients, defined by a baseline EDSS score of ≥3.5 as a proxy for secondary progressive MS, showed a similar reduction in annualised relapse rate compared to patients with an EDSS score below 3.
Pharmacokinetics: Cladribine is a prodrug that has to be phosphorylated intracellularly to become biologically active. Cladribine pharmacokinetics were studied following oral and intravenous administration in MS patients and patients with malignancies, and in
in vitro systems.
Absorption: Following oral administration, cladribine is rapidly absorbed. Administration of 10 mg cladribine resulted in a cladribine mean C
max in the range of 22 to 29 ng/mL and corresponding mean AUC in the range of 80 to 101 ng·h/mL (arithmetic means from various studies).
When oral cladribine was given in fasted state, median T
max was 0.5 h (range 0.5 to 1.5 h). When administered with a high-fat meal, cladribine absorption was delayed (median T
max 1.5 h, range 1 to 3 h) and C
max was reduced by 29% (based on geometric mean), while AUC was unchanged. The bioavailability of 10 mg oral cladribine was approximately 40%.
Distribution: The volume of distribution is large, indicating extensive tissue distribution and intracellular uptake. Studies revealed a mean volume of distribution of cladribine in the range of 480 to 490 L. The plasma protein binding of cladribine is 20%, and independent of plasma concentration.
The distribution of cladribine across biological membranes is facilitated by various transport proteins, including ENT1, CNT3 and BCRP.
In vitro studies indicate that cladribine efflux is only minimally P-gp related. Clinically relevant interactions with inhibitors of P-gp are not expected. The potential consequences of P-gp induction on the bioavailability of cladribine have not been formally studied.
In vitro studies showed negligible transporter-mediated uptake of cladribine into human hepatocytes.
Cladribine has the potential to penetrate the blood brain barrier. A small study in cancer patients has shown a cerebrospinal fluid/plasma concentration ratio of approximately 0.25.
Cladribine and/or its phosphorylated metabolites are substantially accumulated and retained in human lymphocytes.
In vitro, intra- versus extracellular accumulation ratios were found to be around 30 to 40 already 1 hour after cladribine exposure.
Biotransformation: The metabolism of cladribine was studied in MS patients following the administration of a single 10-mg tablet and a single 3-mg intravenous dose. Following both oral and intravenous administration, the parent compound cladribine was the main component present in plasma and urine. The metabolite 2-chloroadenine was a minor metabolite both in plasma and in urine, e.g. accounting only for ≤3% of plasma parent drug exposure after oral administration. Only traces of other metabolites could be found in plasma and in urine.
In hepatic
in vitro systems, negligible metabolism of cladribine was observed (at least 90% was unchanged cladribine).
Cladribine is not a relevant substrate to cytochrome P450 enzymes and does not show significant potential to act as inhibitor of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 and CYP3A4. Inhibition of these enzymes or genetic polymorphisms (e.g. CYP2D6, CYP2C9 or CYP2C19) are not expected to result in clinically significant effects on cladribine pharmacokinetics or exposure. Cladribine has no clinically meaningful inductive effect on CYP1A2, CYP2B6 and CYP3A4 enzymes.
After entering the target cells, cladribine is phosphorylated to cladribine monophosphate (Cd-AMP) by DCK (and also by deoxyguanosine kinase in the mitochondria). Cd-AMP is further phosphorylated to cladribine diphosphate (Cd-ADP) and cladribine triphosphate (Cd-ATP). The dephosphorylation and deactivation of Cd-AMP is catalysed by cytoplasmic 5'-NTase. In a study of the intracellular pharmacokinetics of Cd-AMP and Cd-ATP in patients with chronic myelogenous leukaemia, the levels of Cd-ATP were approximately half of the Cd-AMP levels.
Intracellular half-life of Cd-AMP was 15 h. Intracellular half-life of Cd-ATP was 10 h.
Elimination: Based on pooled population pharmacokinetic data from various studies, the median values for elimination were 22.2 L/h for renal clearance and 23.4 L/h for non-renal clearance. Renal clearance exceeded the glomerular filtration rate, indicating active renal tubular secretion of cladribine.
The non-renal part of the elimination of cladribine (approximately 50%) consists of negligible hepatic metabolism and of extensive intracellular distribution and trapping of the active cladribine principle (Cd-ATP) within the targeted intracellular compartment (i.e. the lymphocytes) and subsequent elimination of intracellular Cd-ATP according to the life-cycle and elimination pathways of these cells.
The estimated terminal half-life for a typical patient from the population pharmacokinetic analysis is approximately 1 day. This however does not result in any drug accumulation after once daily dosing as this half-life only accounts for a small portion of the AUC.
Dose and time dependence: After oral administration of cladribine across a dose range from 3 to 20 mg, C
max and AUC increased in a dose-proportional fashion, suggesting that absorption is not affected by rate- or capacity-limited processes up to a 20 mg oral dose.
No significant accumulation of cladribine concentration in plasma has been observed after repeated dosing. There is no indication that cladribine pharmacokinetics might change in a time-dependent fashion after repeated administration.
Special populations: No studies have been conducted to evaluate the pharmacokinetics of cladribine in elderly or in paediatric MS patients, or in subjects with renal or hepatic impairment.
A population kinetic analysis did not show any effect of age (range 18 to 65 years) or gender on cladribine pharmacokinetics.
Renal impairment: Renal clearance of cladribine was shown to be dependent on creatinine clearance. Based on a population pharmacokinetic analysis including patients with normal renal function and with mild renal impairment, total clearance in patients with mild renal impairment (CL
CR = 60 mL/min) is expected to decrease moderately, leading to an increase in exposure of 25%.
Hepatic impairment: The role of hepatic function for the elimination of cladribine is considered negligible.
Pharmacokinetic interactions: An interaction study in MS patients showed that the bioavailability of 10 mg oral cladribine was not altered when co-administered with pantoprazole.
Toxicology: Preclinical safety data: Non-clinical safety pharmacological and toxicological assessment of cladribine in animal models relevant for the safety assessment of cladribine did not yield significant findings other than those predicted by the pharmacologic mechanism of cladribine. The primary target organs identified in the repeat-dose toxicology studies by parenteral routes (intravenous or subcutaneous) up to 1-year duration in mice and monkeys were the lymphoid and haematopoietic system. Other target organs after longer administration (14 cycles) of cladribine to monkeys by subcutaneous route were the kidneys (karyomegaly of renal tubular epithelium), adrenals (cortex atrophy and decreased vacuolation), gastrointestinal tract (mucosa atrophy) and testes. Effects on the kidneys were also seen in mice.
Mutagenicity: Cladribine is incorporated into DNA strands and inhibits DNA synthesis and repair. Cladribine did not induce gene mutation in bacteria or mammalian cells, but it was clastogenic causing chromosomal damage in mammalian cells
in vitro at a concentration which was 17-fold above the expected clinical C
max.
In vivo clastogenicity in mice was detected at 10 mg/kg, which was the lowest dose tested.
Carcinogenicity: The carcinogenic potential of cladribine was assessed in a long-term 22-month study with subcutaneous administration in mice and in a short-term 26-week study by oral route in transgenic mice.
In the long-term carcinogenicity study in mice, the highest dose used was 10 mg/kg, which was seen to be genotoxic in the mouse micronucleus study (equivalent to approximately 16-fold the expected human exposure in AUC in patients taking the maximum daily dose of 20 mg cladribine). No increased incidence of lymphoproliferative disorders or other tumour types (apart from Harderian gland tumours, predominantly adenomas) was seen in mice. Harderian gland tumours are not considered to be of clinical relevance, as humans do not have comparable anatomical structures.
In the short-term carcinogenicity study in Tg rasH2 mice, no cladribine-related increase in incidence of lymphoproliferative disorders or other tumour types was seen at any dose tested up to 30 mg/kg per day (equivalent to approximately 25-fold the expected human exposure in AUC in patients taking the maximum daily dose of 20 mg cladribine).
Cladribine was also assessed in a 1-year monkey study by the subcutaneous route. No increased incidence in lymphoproliferative disorders and no tumours were seen in this study.
Although cladribine may have a potential for genotoxicity, long-term data in mice and monkeys did not provide any evidence of a relevant increased carcinogenicity risk in humans.
Reproduction toxicity: While there were no effects on female fertility, reproductive function or general performance of offspring, cladribine was shown to be embryolethal when administered to pregnant mice, and the compound was teratogenic in mice (also following treatment of the males only) and rabbits. The observed embryolethal and teratogenic effects are consistent with the pharmacologic mechanisms of cladribine. In a male mouse fertility study, malformed foetuses with agenesis of portions of appendage(s) distal the humerus and/or femur were seen. The incidence of affected mouse foetuses in this study was in the same range of spontaneous incidence of amelia and phocomelia in this strain of mice. However, considering cladribine genotoxicity, male-mediated effects related to potential genetic alteration of differentiating sperm cells cannot be excluded.
Cladribine did not affect the fertility of male mice, but observed testicular effects were reduced testicular weights and increased numbers of non-motile sperm. Testicular degeneration and reversible decrease in spermatozoa with rapid progressive motility were also seen in the monkey. Histologically, testicular degeneration was only seen in one male monkey in a 1-year subcutaneous toxicity study.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image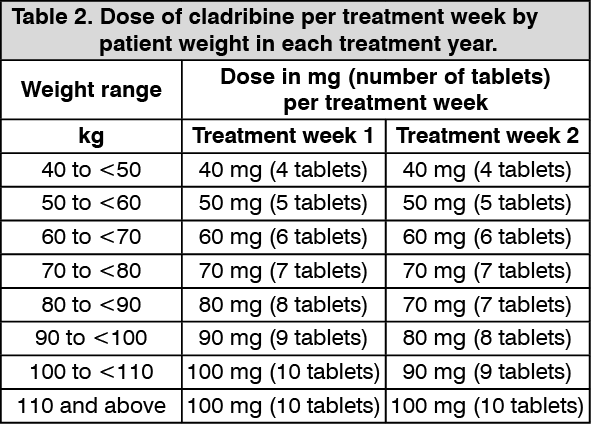 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image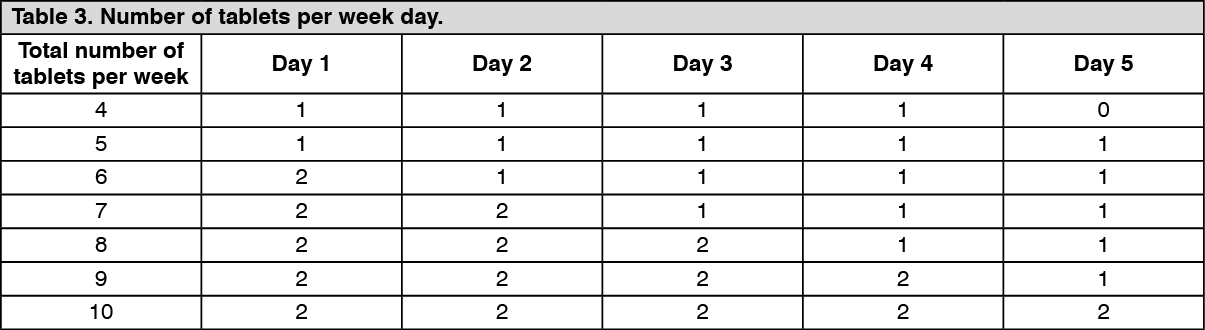 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
