Pharmacology: Mechanism of Action: Sotorasib is an inhibitor of KRASG12C, a tumor-restricted, mutant-oncogenic form of the RAS GTPase, KRAS. Sotorasib forms an irreversible, covalent bond with the unique cysteine of KRASG12C, locking the protein in an inactive state that prevents downstream signaling without affecting wild-type KRAS. Sotorasib blocked KRAS signaling, inhibited cell growth, and promoted apoptosis only in KRAS G12C tumor cell lines. Sotorasib inhibited KRASG12C
in vitro and
in vivo with minimal detectable off-target activity. In mouse tumor xenograft models, sotorasib-treatment led to tumor regressions and prolonged survival, and was associated with anti-tumor immunity in KRAS G12C models.
Pharmacodynamics: Sotorasib exposure-response relationships and the time course of the pharmacodynamic response are unknown.
Cardiac Electrophysiology: At the approved recommended dosage, LUMAKRAS does not cause large mean increases in the QTc interval (>20 msec).
Clinical Studies: The efficacy of LUMAKRAS was demonstrated in a subset of patients enrolled in a single-arm, open-label, multicenter trial (CodeBreaK 100 [NCT03600883]). Eligible patients were required to have locally advanced or metastatic KRAS G12C-mutated NSCLC with disease progression after receiving an immune checkpoint inhibitor and/or platinum-based chemotherapy, an Eastern Cooperative Oncology Group Performance Status (ECOG PS) of 0 or 1, and at least one measurable lesion as defined by Response Evaluation Criteria in Solid Tumors (RECIST v1.1).
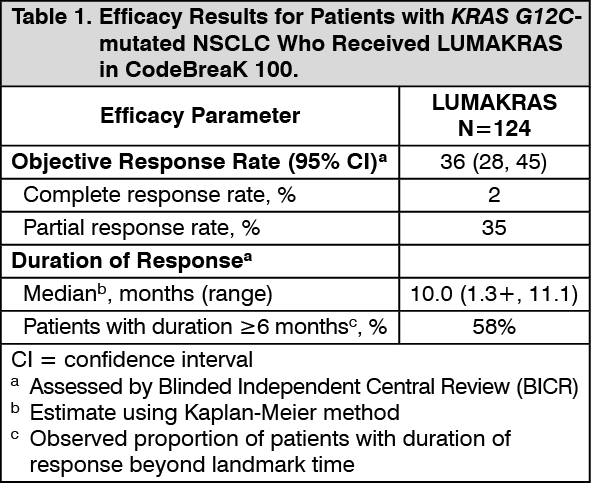
All patients were required to have prospectively identified KRAS G12C-mutated NSCLC in tumor tissue samples by using the QIAGEN therascreen KRAS RGQ PCR Kit performed in a central laboratory. Of 126 total enrolled subjects, 2 (2%) were unevaluable for efficacy analysis due to the absence of radiographically measurable lesions at baseline. Of the 124 patients with KRAS G12C mutations confirmed in tumor tissue, plasma samples from 112 patients were tested retrospectively using the Guardant360 CDx. 78/112 patients (70%) had KRAS G12C mutation identified in plasma specimen, 31/112 patients (28%) did not have KRAS G12C mutation identified in plasma specimen and 3/112 (2%) were unevaluable due to Guardant360 CDx test failure.
A total of 124 patients had at least one measurable lesion at baseline assessed by Blinded Independent Central Review (BICR) according to RECIST v1.1 and were treated with LUMAKRAS 960 mg once daily until disease progression or unacceptable toxicity. The major efficacy outcome measures were objective response rate (ORR) and duration of response (DOR) as evaluated by BICR according to RECIST v1.1.
The baseline demographic and disease characteristics of the study population were: median age 64 years (range: 37 to 80) with 48% ≥65 years and 8% ≥75 years; 50% Female; 82% White, 15% Asian, 2% Black; 70% ECOG PS 1; 96% had stage IV disease; 99% with non-squamous histology; 81% former smokers, 12% current smokers, 5% never smokers. All patients received at least 1 prior line of systemic therapy for metastatic NSCLC; 43% received only 1 prior line of therapy, 35% received 2 prior lines of therapy, 23% received 3 prior lines of therapy; 91% received prior anti-PD-1/PD-L1 immunotherapy, 90% received prior platinum-based chemotherapy, 81% received both platinum-based chemotherapy and anti-PD-1/PD-L1. The sites of known extra-thoracic metastasis included 48% bone, 21% brain, and 21% liver.
Efficacy results are summarized in Table 1. (See Table 1.)
 Click on icon to see table/diagram/image
Pharmacokinetics:
Click on icon to see table/diagram/image
Pharmacokinetics: The pharmacokinetics of sotorasib have been characterized in healthy subjects and in patients with KRAS G12C-mutated solid tumors, including NSCLC. Sotorasib exhibited non-linear, time-dependent, pharmacokinetics over the dose range of 180 mg to 960 mg (0.19 to 1 time the approved recommended dosage) once daily with similar systemic exposure (i.e., AUC
0-24h and C
max) across doses at steady state. Sotorasib systemic exposure was comparable between film-coated tablets and film-coated tablets predispersed in water administered under fasted conditions. Sotorasib plasma concentrations reached steady state within 22 days. No accumulation was observed after repeat LUMAKRAS dosages with a mean accumulation ratio of 0.56 (coefficient of variation (CV): 59%).
Absorption: The median time to sotorasib peak plasma concentration is 1 hour.
Effect of Food: When 960 mg LUMAKRAS was administered with a high-fat, high-calorie meal (containing approximately 800 to 1000 calories with 150, 250, and 500 to 600 calories from protein, carbohydrate and fat, respectively) in patients, sotorasib AUC
0-24h increased by 25% compared to administration under fasted conditions.
Distribution: The sotorasib mean volume of distribution (V
d) at steady state is 211 L (CV: 135%).
In vitro, sotorasib plasma protein binding is 89%.
Elimination: The sotorasib mean terminal elimination half-life is 5 hours (standard deviation (SD): 2). At 960 mg LUMAKRAS once daily, the sotorasib steady state apparent clearance is 26.2 L/hr (CV: 76%).
Metabolism: The main metabolic pathways of sotorasib are non-enzymatic conjugation and oxidative metabolism with CYP3As.
Excretion: After a single dose of radiolabeled sotorasib, 74% of the dose was recovered in feces (53% unchanged) and 6% (1% unchanged) in urine.
Specific Populations: No clinically meaningful differences in the pharmacokinetics of sotorasib were observed based on age (28 to 86 years), sex, race (White, Black and Asian), body weight (36.8 to 157.9 kg), line of therapy, ECOG PS (0, 1), mild and moderate renal impairment (eGFR: ≥30 mL/min/1.73 m
2), or mild hepatic impairment (AST or ALT <2.5 x ULN or total bilirubin <1.5 x ULN). The effect of severe renal impairment or moderate to severe hepatic impairment on sotorasib pharmacokinetics has not been studied.
Drug Interaction Studies: Clinical Studies: Acid-Reducing Agents: Coadministration of repeat doses of omeprazole (PPI) with a single dose of LUMAKRAS decreased sotorasib C
max by 65% and AUC by 57% under fed conditions, and decreased sotorasib C
max by 57% and AUC by 42% under fasted conditions. Coadministration of a single dose of famotidine (H
2 receptor antagonist) given 10 hours prior to and 2 hours after a single dose of LUMAKRAS under fed conditions decreased sotorasib C
max by 35% and AUC by 38%.
Strong CYP3A4 Inducers: Coadministration of repeat doses of rifampin (a strong CYP3A4 inducer) with a single dose of LUMAKRAS decreased sotorasib C
max by 35% and AUC by 51%.
Other Drugs: No clinically meaningful effect on the exposure of sotorasib was observed following coadministration of LUMAKRAS with itraconazole (a combined strong CYP3A4 and P-gp inhibitor) and a single dose of rifampin (an OATP1B1/1B3 inhibitor), or metformin (a MATE1/MATE2-K substrate).
CYP3A4 substrates: Coadministration of LUMAKRAS with midazolam (a sensitive CYP3A4 substrate) decreased midazolam C
max by 48% and AUC by 53%.
P-gp substrates: Coadministration of LUMAKRAS with digoxin (a P-gp substrate) increased digoxin C
max by 91% and AUC by 21%.
MATE1/MATE2-K substrates: No clinically meaningful effect on the exposure of metformin (a MATE1/MATE2-K substrate) was observed following coadministration of LUMAKRAS.
BCRP substrates: Coadministration of LUMAKRAS with rosuvastatin (a BCRP substrate) increased rosuvastatin C
max by 70% and AUC by 34%.
In Vitro Studies: Cytochrome P450 (CYP) Enzymes: Sotorasib may induce CYP2C8, CYP2C9 and CYP2B6. Sotorasib does not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP2D6.
Nonclinical Toxicology: Carcinogenesis, Mutagenesis, Impairment of Fertility: Carcinogenicity studies have not been performed with sotorasib.
Sotorasib was not mutagenic in an
in vitro bacterial reverse mutation (Ames) assay and was not genotoxic in the
in vivo rat micronucleus and comet assays.
Fertility/early embryonic development studies were not conducted with sotorasib. There were no adverse effects on female or male reproductive organs in general toxicology studies conducted in dogs and rats.
Animal Toxicology and/or Pharmacology: In rats, renal toxicity including minimal to marked histologic tubular degeneration/necrosis and increased kidney weight, urea nitrogen, creatinine, and urinary biomarkers of renal tubular injury were present at doses resulting in exposures approximately ≥0.5 times the human AUC at the clinical dose of 960 mg. Increases in cysteine S-conjugate β-lyase pathway metabolism in the rat kidney compared to human may make rats more susceptible to renal toxicity due to local formation of a putative sulfur-containing metabolite than humans.
In the 3-month toxicology study in dogs, sotorasib induced findings in the liver (centrilobular hepatocellular hypertrophy), pituitary gland (hypertrophy of basophils), and thyroid gland (marked follicular cell atrophy, moderate to marked colloid depletion, and follicular cell hypertrophy) at exposures approximately 0.4 times the human exposure based on AUC at the clinical dose of 960 mg. These findings may be due to an adaptive response to hepatocellular enzyme induction and subsequent reduced thyroid hormone levels (i.e., secondary hypothyroidism). Although thyroid levels were not measured in dogs, induction of uridine diphosphate glucuronosyltransferase known to be involved in thyroid hormone metabolism was confirmed in the
in vitro dog hepatocyte assay.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
