The following clinically significant adverse reactions are described in other sections of the monographs: Cardiac Toxicities [see Cardiac Toxicities under Precautions].
Acute Renal Failure [see Acute Renal Failure under Precautions].
Tumor Lysis Syndrome [see Tumor Lysis Syndrome under Precautions].
Pulmonary Toxicity [see Pulmonary Toxicity under Precautions].
Pulmonary Hypertension [see Pulmonary Hypertension under Precautions].
Dyspnea [see Dyspnea under Precautions].
Hypertension [see Hypertension under Precautions].
Venous Thrombosis [see Venous Thrombosis under Precautions].
Infusion-Related Reactions [see Infusion-Related Reactions under Precautions].
Hemorrhage [see Hemorrhage under Precautions].
Thrombocytopenia [see Thrombocytopenia under Precautions].
Hepatic Toxicity and Hepatic Failure [see Hepatic Toxicity and Hepatic Failure under Precautions].
Thrombotic Microangiopathy [see Thrombotic Microangiopathy under Precautions].
Posterior Reversible Encephalopathy Syndrome [see Posterior Reversible Encephalopathy Syndrome under Precautions].
Progressive Multifocal Leukoencephalopathy [see Progressive Multifocal Leukoencephalopathy under Precautions].
Increased Fatal and Serious Toxicities in Combination with Melphalan and Prednisone in Newly Diagnosed Transplant-Ineligible Patients [see Increased Fatal and Serious Toxicities in Combination with Melphalan and Prednisone in Newly Diagnosed Transplant-Ineligible Patients under Precautions].
Clinical Trials Experience: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug, and may not reflect the rates observed in medical practice.
The pooled safety population described previously in Precautions reflect exposure to Kyprolis in 1789 patients administered in combination with other drugs in ASPIRE, ENDEAVOR, A.R.R.O.W., and CANDOR. The most common adverse reactions occuring in at least 20% of patients who received Kyprolis in combination were anemia, diarrhea, fatigue, hypertension, pyrexia, upper respiratory tract infection, thrombocytopenia, cough, dyspnea, and insomnia.
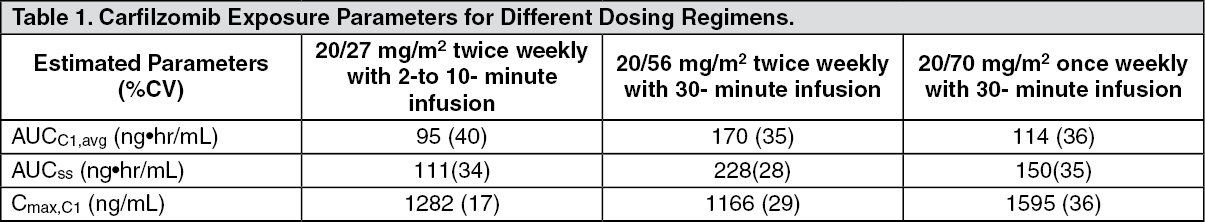
Kyprolis in Combination with Lenalidomide and Dexamethasone: The safety of Kyprolis 20/27 mg/m
2 twice weekly in combination with lenalidomide and dexamethasone (KRd) was evaluated in ASPIRE [Pharmacology: Clinical Studies: In Combination with Lenalidomide and Dexamethasone for Relapsed or Refractory Multiple Myeloma under Actions]. The median number of cycles initiated was 22 cycles for the KRd arm and 14 cycles for the Rd arm.
Deaths due to adverse reactions within 30 days of the last dose of any therapy in the KRd arm occurred in 45/392 (12%) patients compared with 42/389 (11%) patients who died due to adverse reactions within 30 days of the last dose af any Rd therapy. The most frequent cause of deaths occurring in patients (%) in the two arms (KRd
versus Rd) included infection 12 (3%)
versus 11 (3%), cardiac 10 (3%)
versus 9 (2%), and other adverse reactions 23 (6%)
versus 22 (6%).
Serious adverse reactions were reported in 65% of the patients in the KRd arm and 57% of the patients in the Rd arm. The most frequent serious adverse reactions reported in the KRd arm as compared with the Rd arm were pneumonia (17%
versus 13%), respiratory tract infection (4%
versus 2%), pyrexia (4%
versus 3%), and pulmonary embolism (3%
versus 2%).
Discontinuation due to any adverse reactions occurred in 33% in the KRd arm
versus 30% in the Rd arm. Adverse reactions leading to discontinuation of Kyprolis occurred in 12% of patients and the most common reactions included pneumonia (1%), myocardial infarction (0.8%), and upper respiratory tract infection (0.8%). The incidence of cardiac failure events was 7% in the KRd arm versus 4% in the Rd arm.
Table 21 summarizes the adverse reactions in the first 12 cycles in ASPIRE. (See Table 21.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
There were 274 (70%) patients in the KRd arm who received treatment beyond Cycle 12. There were no new clinically relevant adverse reactions that emerged in the later treatment cycles.
Adverse Reactions Occurring at a Frequency of <10%: Blood and lymphatic system disorders: febrile neutropenia, lymphopenia.
Cardiac disorders: cardiac arrest, cardiac failure, cardiac failure congestive, myocardial infarction, myocardial ischemia, pericardial effusion.
Ear and labyrinth disorders: deafness, tinnitus.
Eye disorders: cataract, vision blurred.
Gastrointestinal disorders: abdominal pain, abdominal pain upper, dyspepsia, gastrointestinal hemorrhage, toothache.
General disorders and administration site conditions: chills, infusion site reaction, multi-organ failure, pain.
Infections and infestations:
Clostridium difficile colitis, influenza, lung infection, rhinitis, sepsis, urinary tract infection, viral infection.
Metabolism and nutrition disorders: dehydration, hyperkalemia, hyperuricemia, hypoalbuminemia, hyponatremia, tumor lysis syndrome.
Musculoskeletal and connective tissue disorders: muscular weakness, myalgia.
Nervous system disorders: hypoesthesia, intracranial hemorrhage, paresthesia.
Psychiatric disorders: anxiety, delirium.
Renal and urinary disorders: renal failure, renal failure acute, renal impairment.
Respiratory, thoracic and mediastinal disorders: dysphonia, epistaxis, oropharyngeal pain, pulmonary embolism, pulmonary edema, pulmonary hemorrhage.
Skin and subcutaneous tissue disorders: erythema, hyperhidrosis, pruritus.
Vascular disorders: deep vein thrombosis, hemorrhage, hypotension.
Grade 3 and higher adverse reactions that occurred during Cycles 1-12 with a substantial difference (≥ 2%) between the two arms were neutropenia, thrombocytopenia, hypokalemia, and hypophosphatemia.
Table 22 describes Grade 3-4 laboratory abnormalities reported in ASPIRE. (See Table 22.)
Click on icon to see table/diagram/image
Kyprolis in Combination with Dexamethasone: The safety of Kyprolis in combination with dexamethasone was evaluated in two open-label, randomized trials (ENDEAVOR and A.R.R.O.W.).
ENDEAVOR: The safety of Kyprolis 20/56 mg/m
2 twice weekly in combination with dexamethasone (Kd) was evaluated in ENDEAVOR [Pharmacology: Clinical Studies: In Combination with Dexamethasone for Relapsed or Refractory Multiple Myeloma under Actions].
Deaths due to adverse reactions within 30 days of last study treatment occurred in 32/463 (7%) patients in the Kd arm and 21/456 (5%) patients in the Vd arm. The causes of death occurring in patients (%) in the two arms (Kd
versus Vd) included cardiac 4 (1%)
versus 5 (1%), infections 8 (2%)
versus 8 (2%), disease progression 7 (2%)
versus 4 (1%), pulmonary 3 (1%)
versus 2 (< 1%), renal 1 (< 1%)
versus 0 (0%), and other adverse reactions 9 (2%)
versus 2 (< 1%).
Serious adverse reactions were reported in 59% of the patients in the Kd arm and 40% of the patients in the Vd arm. In both arms, pneumonia was the most frequently reported serious adverse reaction (8%
versus 9%).
Discontinuation due to any adverse reaction occurred in 29% in the Kd arm versus 26% in the Vd arm. The most frequent adverse reaction leading to discontinuation was cardiac failure in the Kd arm (n = 8, 2%) and peripheral neuropathy in the Vd arm (n = 22, 5%). The incidence of cardiac failure events was 11% in the Kd arm versus 3% in the Vd arm.
Adverse reactions in the first 6 months of therapy that occurred at a rate of 10% or greater in the Kd arm are presented in Table 23. (See Table 23.)
Click on icon to see table/diagram/image
The event rate of ≥ Grade 2 peripheral neuropathy in the Kd arm was 7% (95% CI: 5, 9)
versus 35% (95% CI: 31, 39) in the Vd arm.
Adverse Reactions Occurring at a Frequency of < 10%: Blood and lymphatic system disorders: febrile neutropenia, leukopenia, lymphopenia, neutropenia, thrombotic microangiopathy, thrombotic thrombocytopenic purpura.
Cardiac disorders: atrial fibrillation, cardiac arrest, cardiac failure, cardiac failure congestive, myocardial infarction, myocardial ischemia, palpitations, tachycardia.
Ear and labyrinth disorders: tinnitus.
Eye disorders: cataract, vision blurred.
Gastrointestinal disorders: abdominal pain, abdominal pain upper, dyspepsia, gastrointestinal hemorrhage, toothache.
General disorders and administration site conditions: chest pain, chills, influenza like illness, infusion site reactions (including inflammation, pain, and erythema), malaise, pain.
Hepatobiliary disorders: cholestasis, hepatic failure, hyperbilirubinemia.
Immune system disorders: drug hypersensitivity.
Infections and infestations: bronchopneumonia, gastroenteritis, influenza, lung infection, nasopharyngitis, pneumonia, rhinitis, sepsis, urinary tract infection, viral infection.
Metabolism and nutrition disorders: decreased appetite, dehydration, hypercalcemia, hyperkalemia, hyperuricemia, hypoalbuminemia, hypocalcemia, hypomagnesemia, hyponatremia, hypophosphatemia, tumor lysis syndrome.
Musculoskeletal and connective tissue disorders: muscular weakness, musculoskeletal chest pain, musculoskeletal pain, myalgia.
Nervous system disorders: cerebrovascular accident, dizziness, hypoesthesia, paresthesia, posterior reversible encephalopathy syndrome.
Psychiatric disorders: anxiety.
Renal and urinary disorders: renal failure, renal failure acute, renal impairment.
Respiratory, thoracic and mediastinal disorders: acute respiratory distress syndrome, dysphonia, epistaxis, interstitial lung disease, oropharyngeal pain, pneumonitis, pulmonary embolism, pulmonary edema, pulmonary hypertension, wheezing.
Skin and subcutaneous tissue disorders: erythema, hyperhidrosis, pruritus, rash.
Vascular disorders: deep vein thrombosis, flushing, hypotension.
Table 24 describes Grade 3-4 laboratory abnormalities reported at a rate of ≥ 10% in the Kd arm. (See Table 24.)
Click on icon to see table/diagram/image
A.R.R.O.W.: The safety of Kyprolis in combination with dexamethasone was evaluated in A.R.R.O.W. [Pharmacology: Clinical Studies under Actions]. Patients received treatment for a median duration of 38 weeks in the Kd 20/70 mg/m2 arm once weekly and 29.1 weeks in the Kd 20/27 mg/m
2 twice weekly arm. The safety profile for the once weekly Kd 20/70 mg/m
2 regimen was similar to the twice weekly Kd 20/27 mg/m
2 regimen.
Deaths due to adverse reactions within 30 days of last study treatment occurred in 22/238 (9%) patients in the Kd 20/70 mg/m
2 arm and 18/235 (8%) patients in the Kd 20/27 mg/m
2 arm. The most frequent fatal adverse reactions occurring in patients (%) in the two arms (once weekly Kd 20/70 mg/m
2 versus twice weekly Kd 20/27 mg/m
2) were sepsis 2 (< 1%) versus 2 (< 1%), septic shock 2 (< 1%)
versus 1 (< 1%), and infection 2 (< 1%) versus 0 (0%).
Serious adverse reactions were reported in 43% of the patients in the Kd 20/70 mg/m
2 arm and 41% of the patients in the Kd 20/27 mg/m
2 arm. In both arms, pneumonia was the most frequently reported serious adverse reaction (8% versus 7%).
Discontinuation due to any adverse reaction occurred in 13% in the Kd 20/70 mg/m
2 arm versus 12% in the Kd 20/27 mg/m
2 arm. The most frequent adverse reaction leading to discontinuation was acute kidney injury (2%
versus 2%). The incidence of cardiac failure events was 3.8% in the once weekly Kd 20/70 mg/m
2 arm
versus 5.1% in the twice weekly Kd 20/27 mg/m
2 arm.
Adverse reactions that occurred at a rate of 10% or greater in either Kd arm is presented in Table 25. (See Table 25.)
Click on icon to see table/diagram/image
Adverse Reactions Occurring at a Frequency of < 10%: Blood and lymphatic system disorders: febrile neutropenia, leukopenia, lymphopenia, neutropenia, thrombotic microangiopathy.
Cardiac disorders: atrial fibrillation, cardiac arrest, cardiac failure, cardiac failure congestive, myocardial infarction, myocardial ischemia, palpitations, pericardial effusion, tachycardia.
Ear and labyrinth disorders: tinnitus.
Eye disorders: cataract, vision blurred.
Gastrointestinal disorders: abdominal pain, abdominal pain upper, constipation, dyspepsia, toothache, vomiting.
General disorders and administration site conditions: chest pain, chills, influenza like illness, infusion site reactions (including inflammation, pain, and erythema), malaise, pain.
Hepatobiliary disorders: cholestasis, hepatic failure, hyperbilirubinemia.
Infections and infestations: Clostridium difficile colitis, gastroenteritis, influenza, lung infection, nasopharyngitis, rhinitis, sepsis, septic shock, urinary tract infection, viral infection.
Metabolism and nutrition disorders: decreased appetite, dehydration, hypercalcemia, hyperglycemia, hyperkalemia, hyperuricemia, hypoalbuminemia, hypocalcemia, hypomagnesemia, hyponatremia, hypophosphatemia, tumor lysis syndrome.
Musculoskeletal and connective tissue disorders: muscle spasms, muscular weakness, musculoskeletal chest pain, musculoskeletal pain, myalgia.
Nervous system disorders: cerebrovascular accident, dizziness, paresthesia, peripheral neuropathy.
Psychiatric disorders: anxiety, delirium.
Renal and urinary disorders: acute kidney injury, renal failure, renal impairment.
Respiratory, thoracic and mediastinal disorders: acute respiratory distress syndrome, dysphonia, epistaxis, interstitial lung disease, oropharyngeal pain, pneumonitis, pulmonary hemorrhage, pulmonary embolism, pulmonary hypertension, pulmonary edema, wheezing.
Skin and subcutaneous tissue disorders: erythema, hyperhidrosis, pruritus, rash.
Vascular disorders: deep vein thrombosis, flushing, hypotension.
Kyprolis in Combination with Intravenous Daratumumab and Dexamethasone: The safety of Kyprolis in combination with intravenous daratumumab and dexamethasone was evaluated in two trials (CANDOR and EQUULEUS).
CANDOR: The safety of Kyprolis 20/56 mg/m
2 twice weekly in combination with intravenous daratumumab and dexamethasone (DKd) was evaluated in CANDOR [see Pharmacology: Clinical Studies: In Combination with Intravenous Daratumumab and Dexamethasone for Relapsed or Refractory Multiple Myeloma under Actions]. Patients received Kyprolis for a median duration of 58 weeks in the DKd arm and 40 weeks in the Kd arm.
Serious adverse reactions were reported in 56% of the patients in the DKd arm and 46% of the patients in the Kd arm. The most frequent serious adverse reactions reported in the DKd arm as compared with the Kd arm were pneumonia (14%
versus 9%), pyrexia (4.2%
versus 2.0%), influenza (3.9%
versus 1.3%), sepsis (3.9%
versus 1.3%), anemia (2.3%
versus 0.7%), bronchitis (1.9%
versus 0%) and diarrhea (1.6%
versus 0%). Fatal adverse reactions within 30 days of the last dose of any study treatment occurred in 10% of 308 patients in the DKd arm compared with 5% of 153 patients in the Kd arm. The most frequent fatal adverse reaction (DKd
versus Kd) was infection 4.5% versus 2.6%.
Permanent discontinuation due to an adverse reaction in patients who received Kyprolis occurred in 21% of patients in the DKd arm
versus 22% in the Kd arm. The most frequent adverse reactions leading to discontinuation of Kyprolis were cardiac failure (1.9%) and fatigue (1.9%) in the DKd arm and cardiac failure (2.0%), hypertension (2.0%) and acute kidney injury (2.0%) in the Kd arm. Interruption of Kyprolis due to adverse reactions occurred in 71% of patients in DKd arm versus 63% in the Kd arm. Dose reduction of Kyprolis due to adverse reactions occurred in 25% of patients in DKd arm
versus 20% in the Kd arm.
Infusion-related reactions that occurred following the first Kyprolis dose was 13% in the DKd arm versus 1% in the Kd arm.
Table 26 summarizes the adverse reactions in CANDOR. (See Table 26.)
Click on icon to see table/diagram/image
Adverse Reactions Occurring at a Frequency of < 15%: Blood and lymphatic system disorders: febrile neutropenia, thrombotic thrombocytopenic purpura.
Cardiac disorders: atrial fibrillation, cardiac arrest, cardiac failure, cardiomyopathy, myocardial infarction, myocardial ischemia, tachycardia.
Eye disorders: cataract.
Gastrointestinal disorders: abdominal pain, gastrointestinal hemorrhage.
General disorders and administration site conditions: chest pain, malaise.
Infections: gastroenteritis, influenza, lung infection, nasopharyngitis, sepsis, septic shock, urinary tract infection, viral infection.
Investigations: alanine aminotransferase increased, blood creatinine increased, C-reactive protein increased, ejection fraction decreased.
Metabolism and nutrition disorders: dehydration, hyperglycemia, hyperkalemia, hypokalemia, hyponatremia, tumor lysis syndrome.
Musculoskeletal and connective tissue disorders: pain in extremity.
Nervous system disorders: cerebrovascular accident, intracranial hemorrhage, posterior reversible encephalopathy syndrome, peripheral neuropathy.
Psychiatric disorders: anxiety.
Renal and urinary disorders: acute kidney injury, renal failure, renal impairment.
Respiratory, thoracic and mediastinal disorders: acute respiratory failure, epistaxis, interstitial lung disease, pneumonitis, pulmonary embolism, pulmonary hypertension, pulmonary edema.
Skin and subcutaneous tissue disorders: rash.
Vascular disorders: deep vein thrombosis, hypertensive crisis.
EQUULEUS: The safety of Kyprolis 20/70 mg/m2 once weekly in combination with daratumumab and dexamethasone (DKd) was evaluated in EQUULEUS [see Pharmacology: Pharmacokinetics under Actions]. Patients received Kyprolis for a median duration of 66 weeks.
Serious adverse reactions were reported in 48% of patients. The most frequent serious adverse reactions reported were pneumonia (4.7%), upper respiratory tract infection (4.7%), basal cell carcinoma (4.7%), influenza (3.5%), general physical health deterioration (3.5%) and hypercalcemia (3.5%). Fatal adverse reactions within 30 days of the last dose of any study treatment occurred in 3.5% of patients who died of general physical health deterioration, multi-organ failure secondary to pulmonary aspergillosis, and disease progression.
Discontinuation of Kyprolis occurred in 19% of patients. The most frequent adverse reaction leading to discontinuation was asthenia (2%). Interruption of Kyprolis due to adverse reactions occurred in 77% of patients. Dose reduction of Kyprolis due to adverse reactions occurred in 31% of patients in DKd.
Infusion-related reactions that occurred following the first Kyprolis dose was 11%. Pulmonary hypertension adverse reactions were reported in 4.7% of patients in EQUULEUS.
Table 27 summarizes the adverse reactions in EQUULEUS. (See Table 27.)
Click on icon to see table/diagram/image
Adverse Reactions Occurring at a Frequency of < 15%: Blood and lymphatic system disorders: febrile neutropenia, thrombotic microangiopathy.
Cardiac disorders: cardiac failure, myocardial ischemia.
Gastrointestinal disorders: abdominal pain.
General disorders and administration site conditions: multiple organ dysfunction syndrome.
Infections: pneumonia, sepsis, septic shock.
Metabolism and nutrition disorders: dehydration, hypercalcemia.
Renal and urinary disorders: acute kidney injury, renal failure, renal impairment.
Respiratory, thoracic and mediastinal disorders: pulmonary embolism, pulmonary hypertension.
Vascular disorders: hypotension.
Postmarketing Experience: The following adverse reactions have been identified during post approval use of Kyprolis. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure: hemolytic uremic syndrome (HUS), hepatitis B virus reactivation, gastrointestinal perforation, pericarditis, and cytomegalovirus infection including chorioretinitis, pneumonitis, enterocolitis, and viremia.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image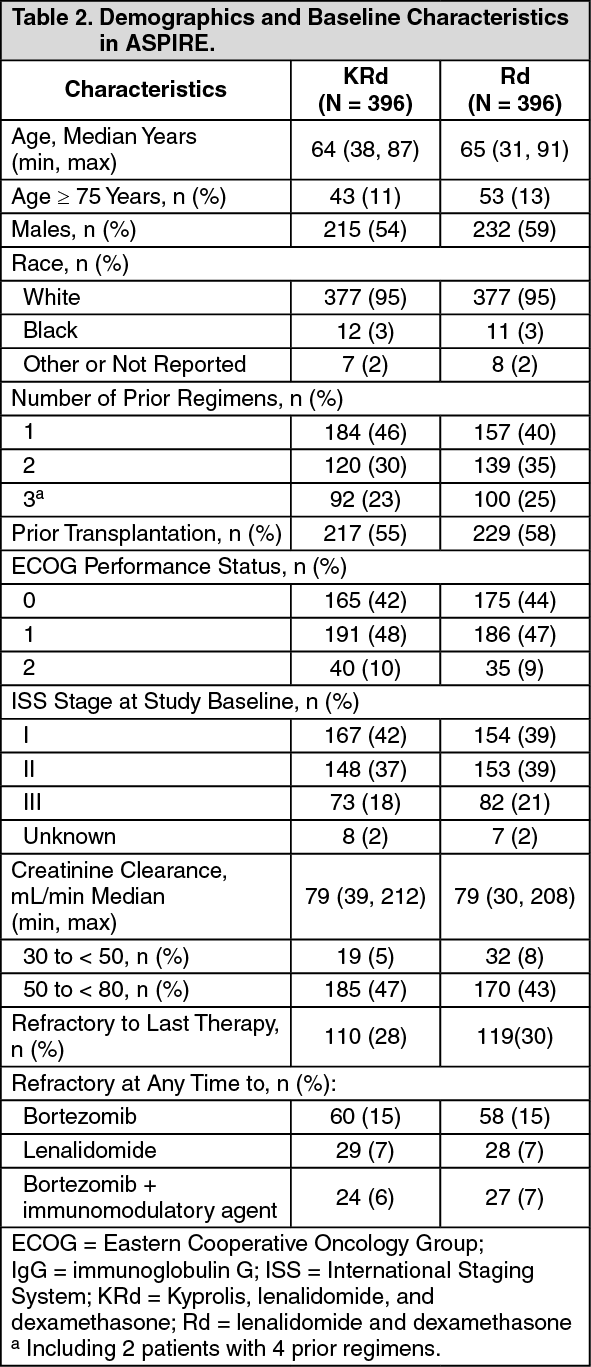 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image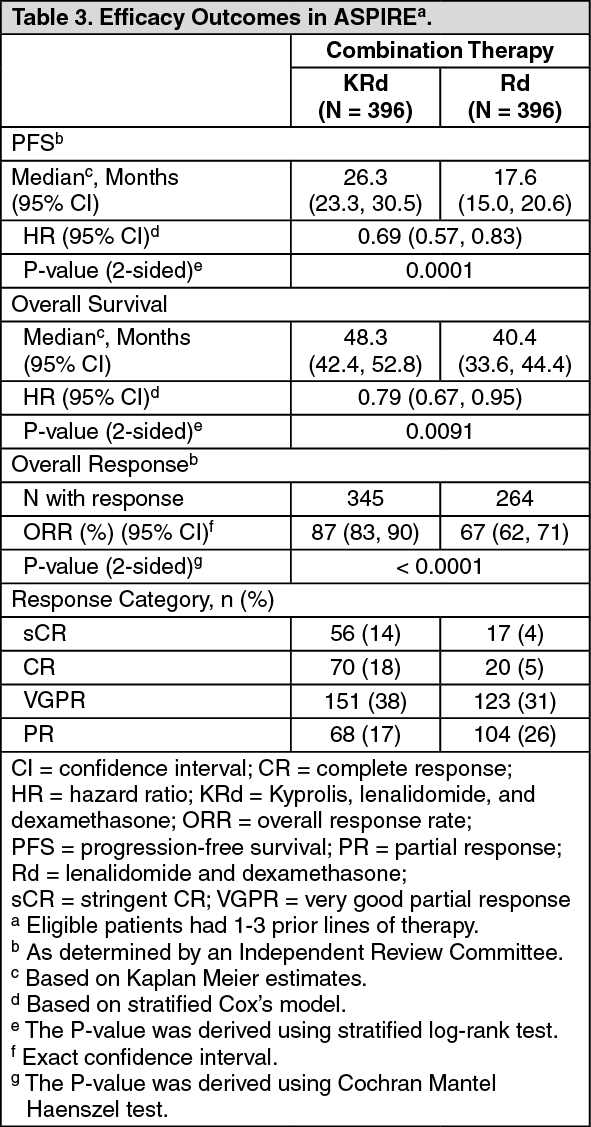 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image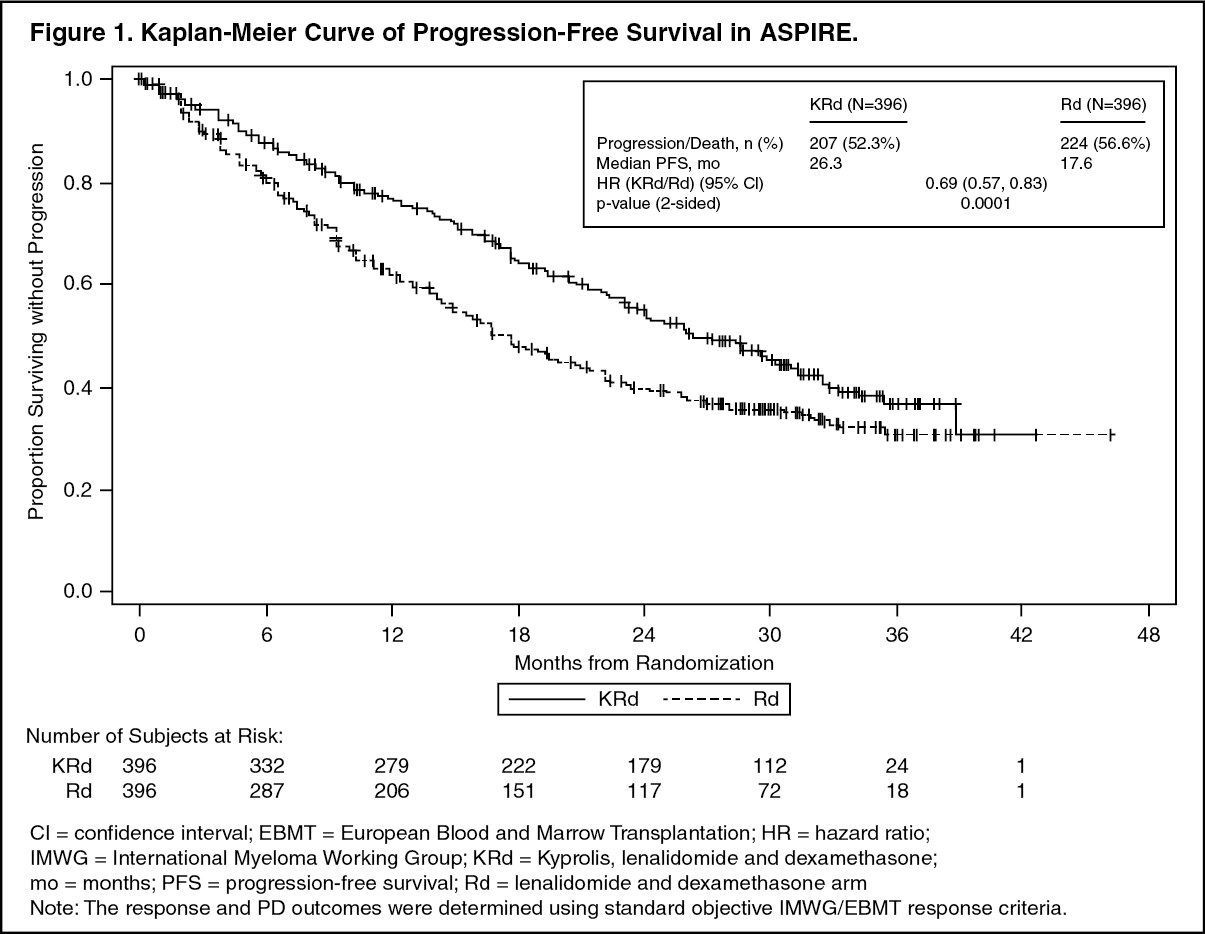 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image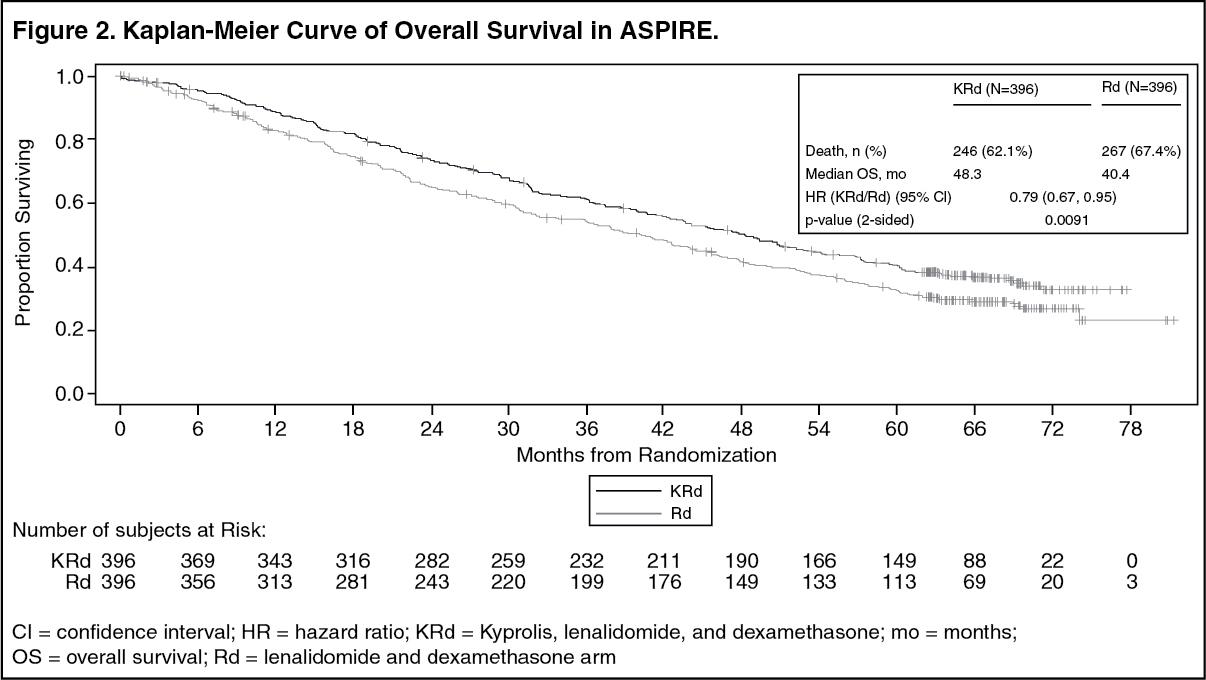 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image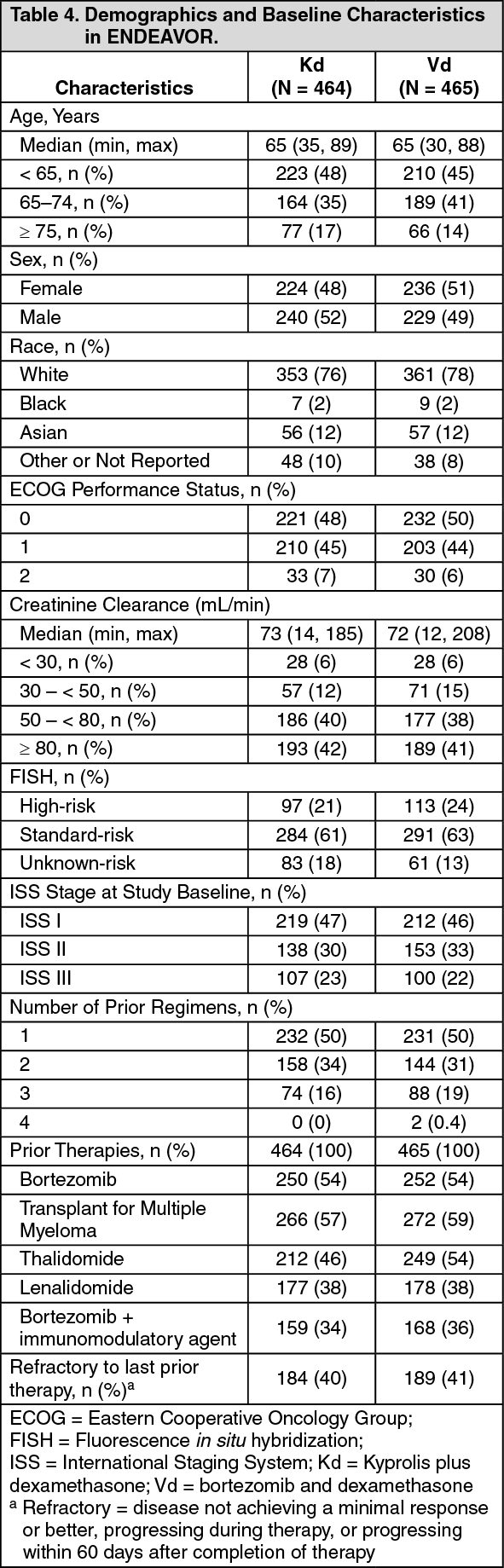 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image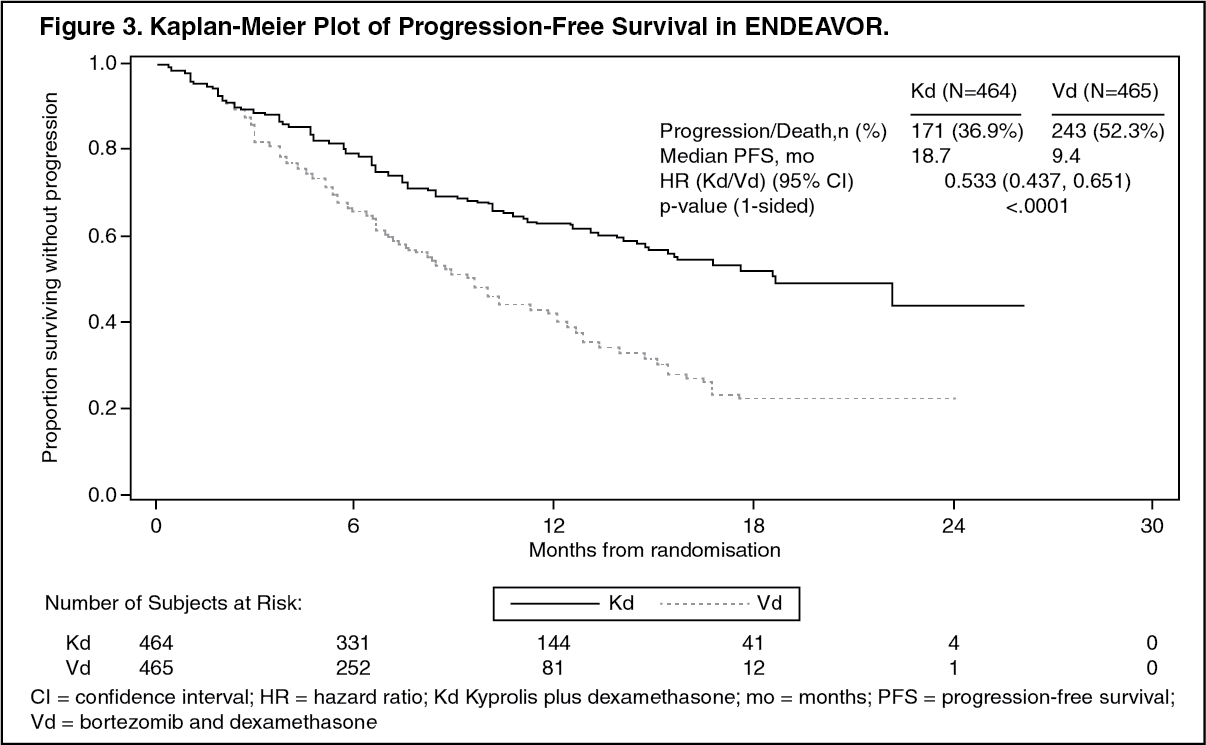 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image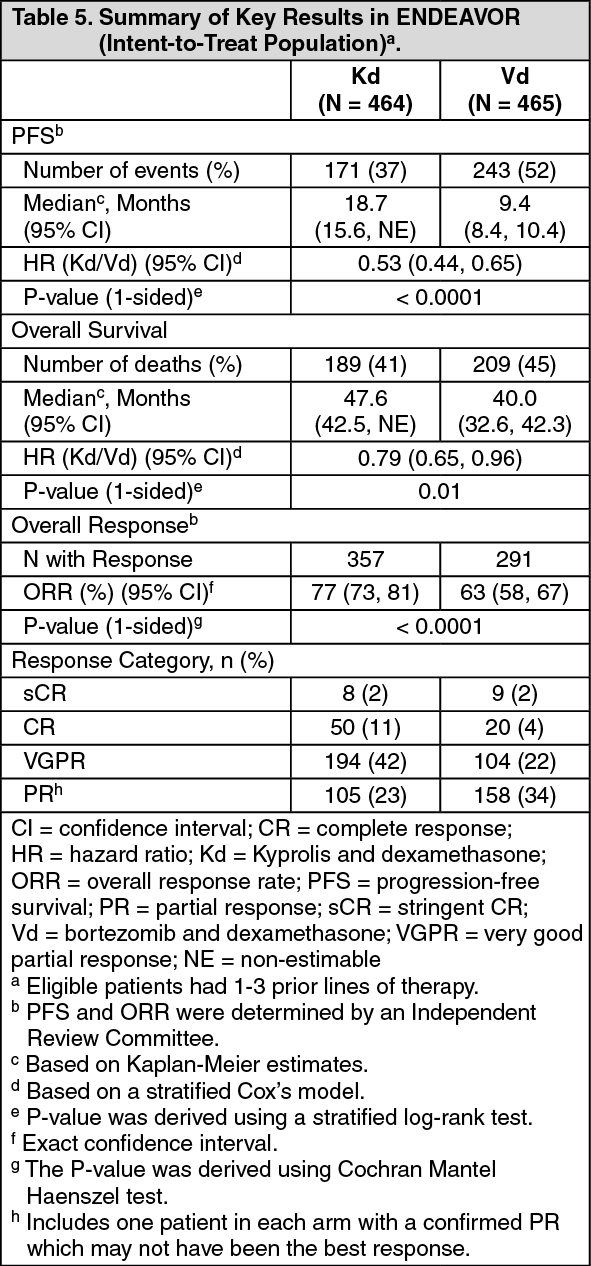 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image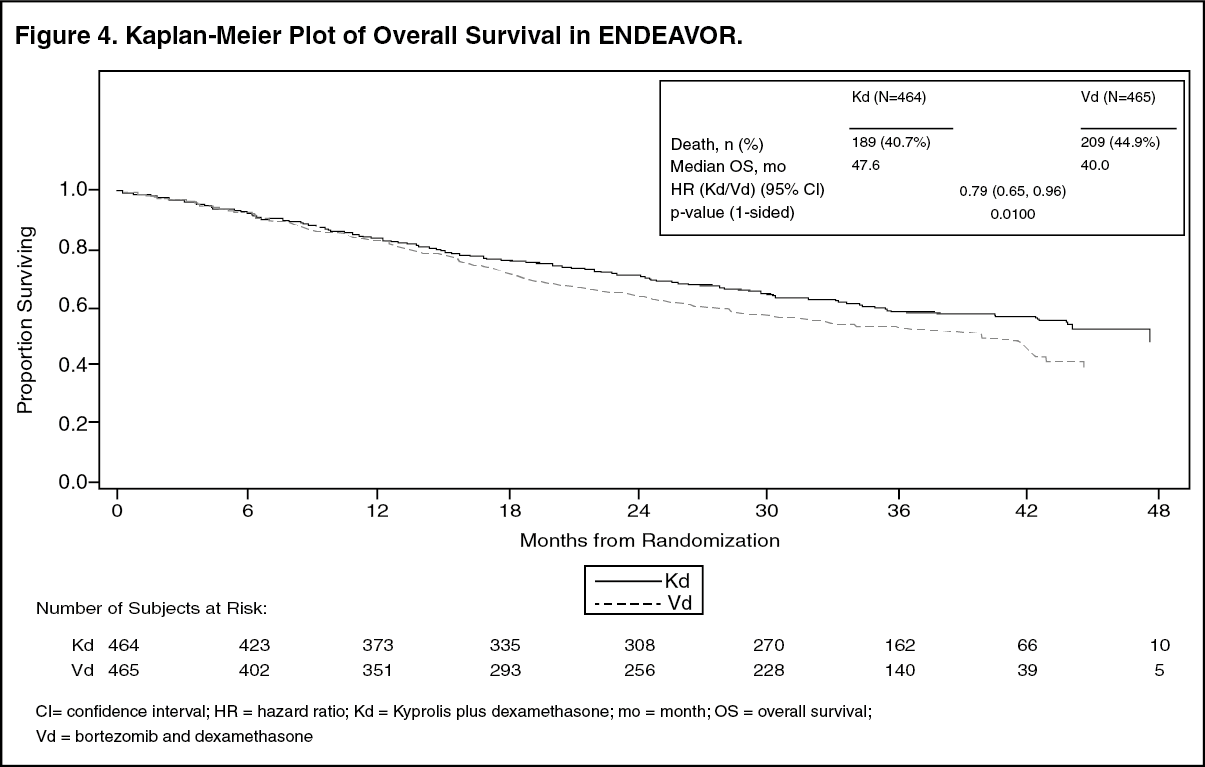 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image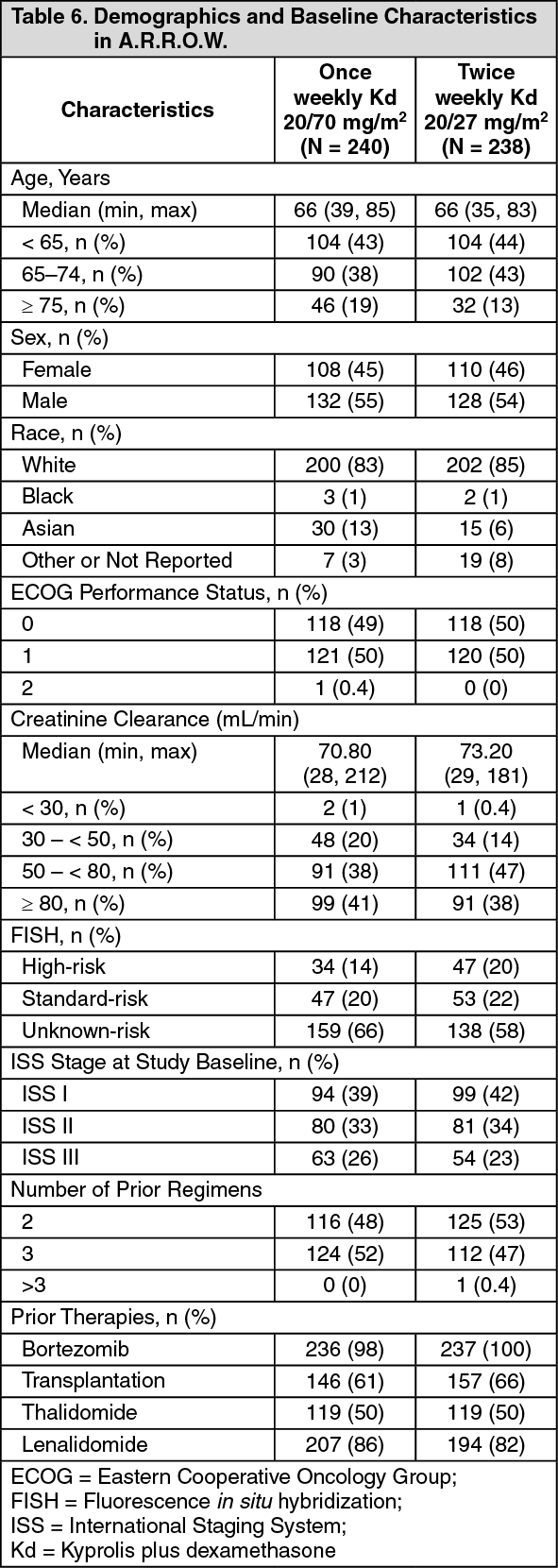 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image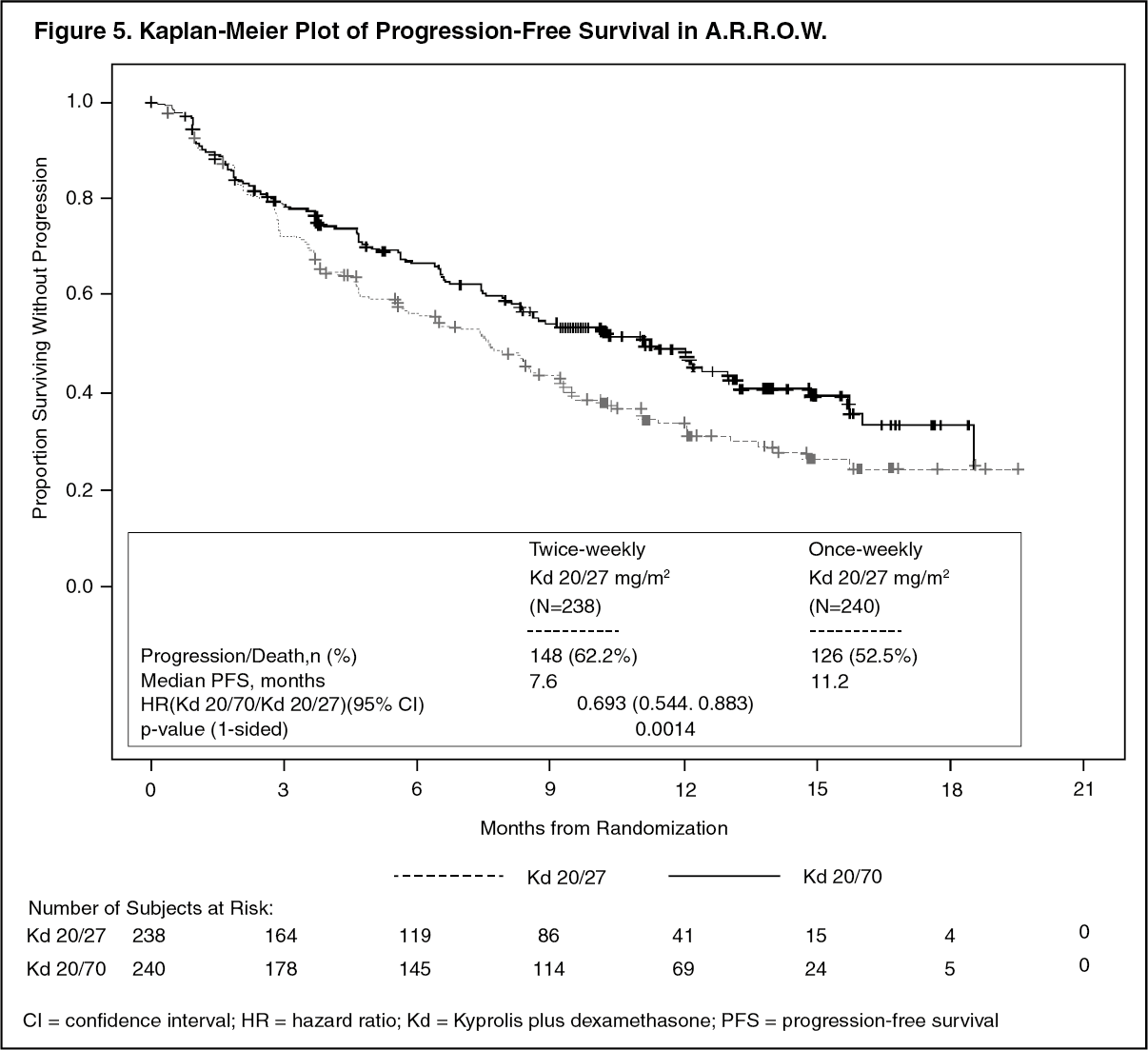 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image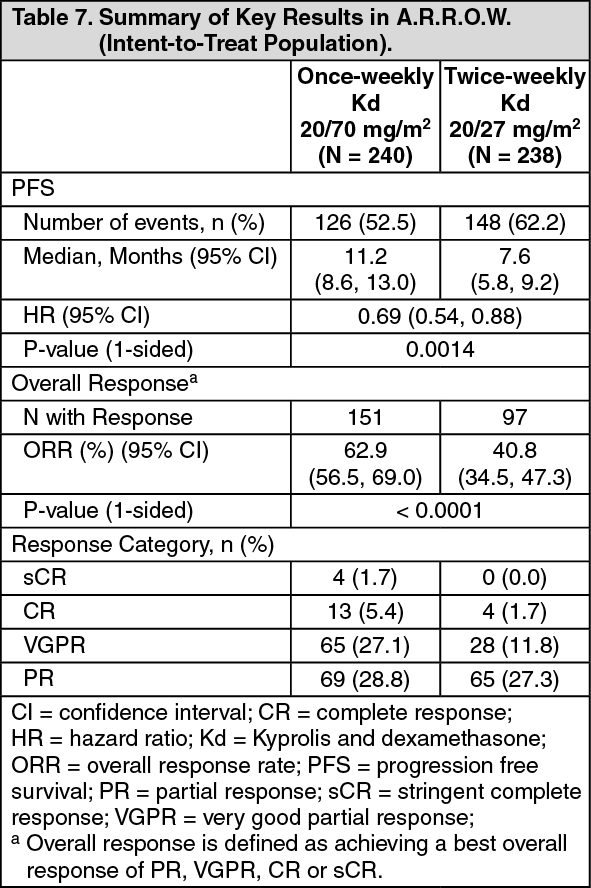 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image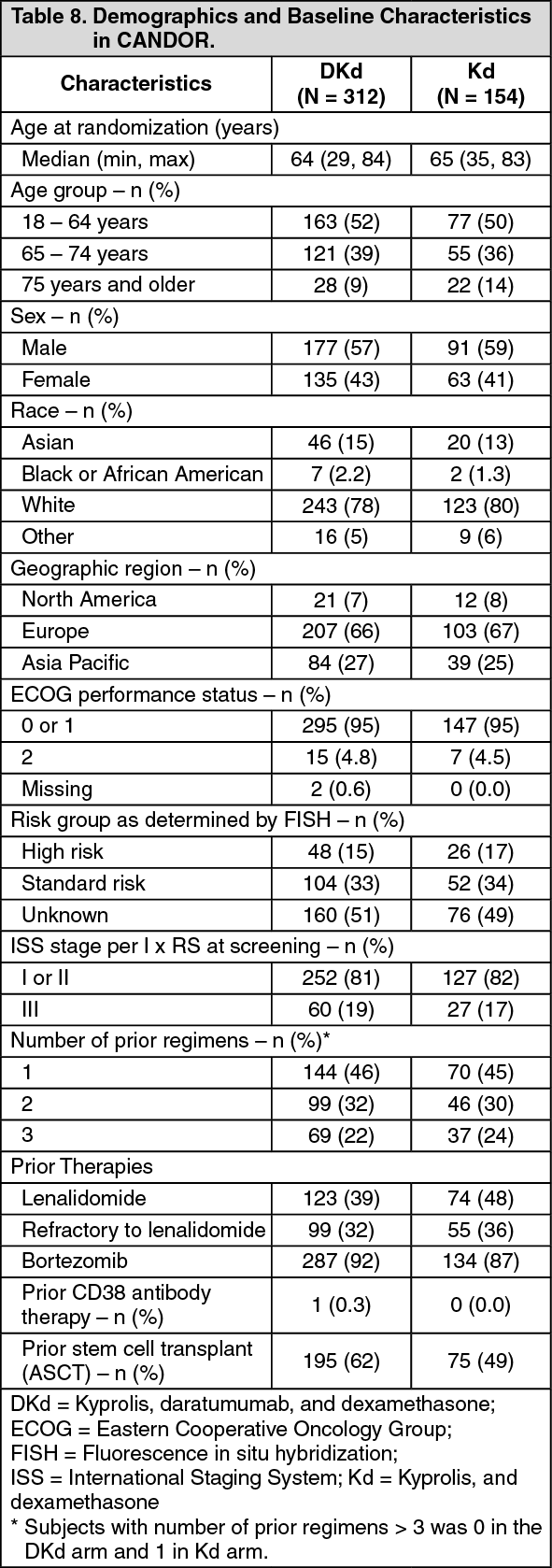 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image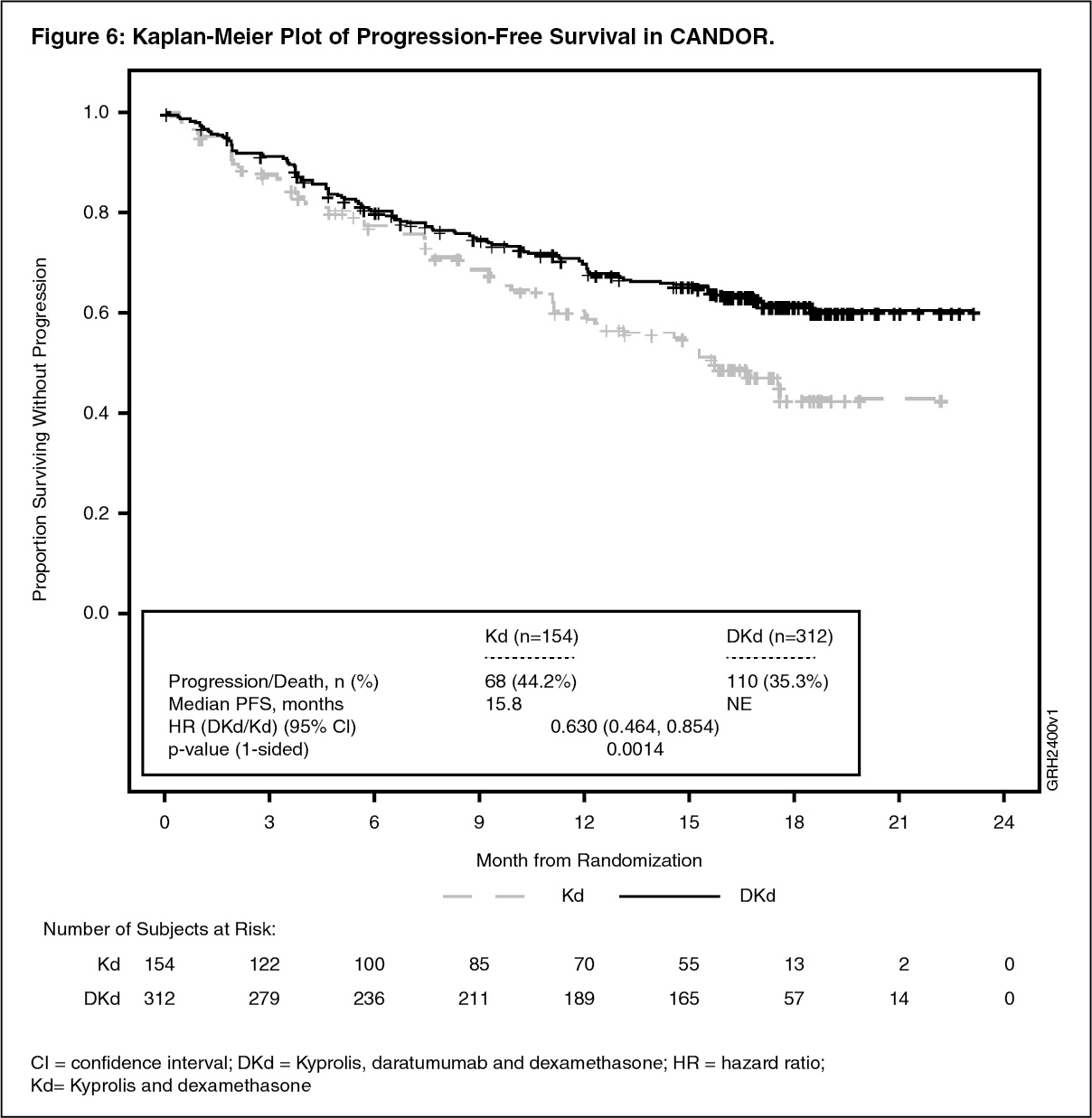 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image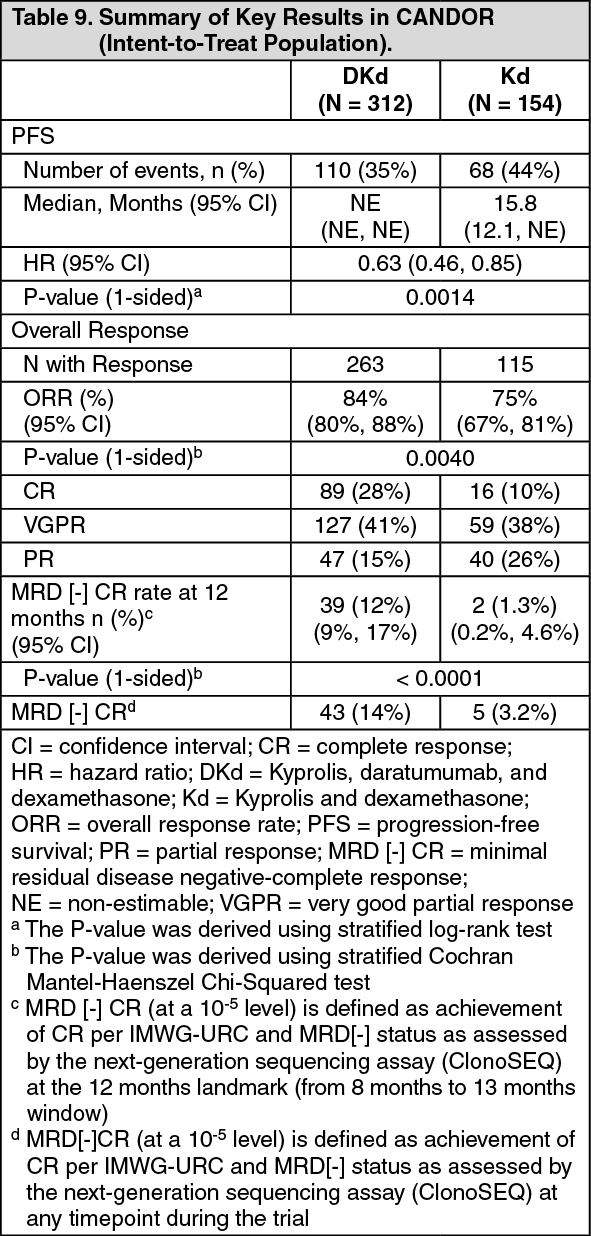 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image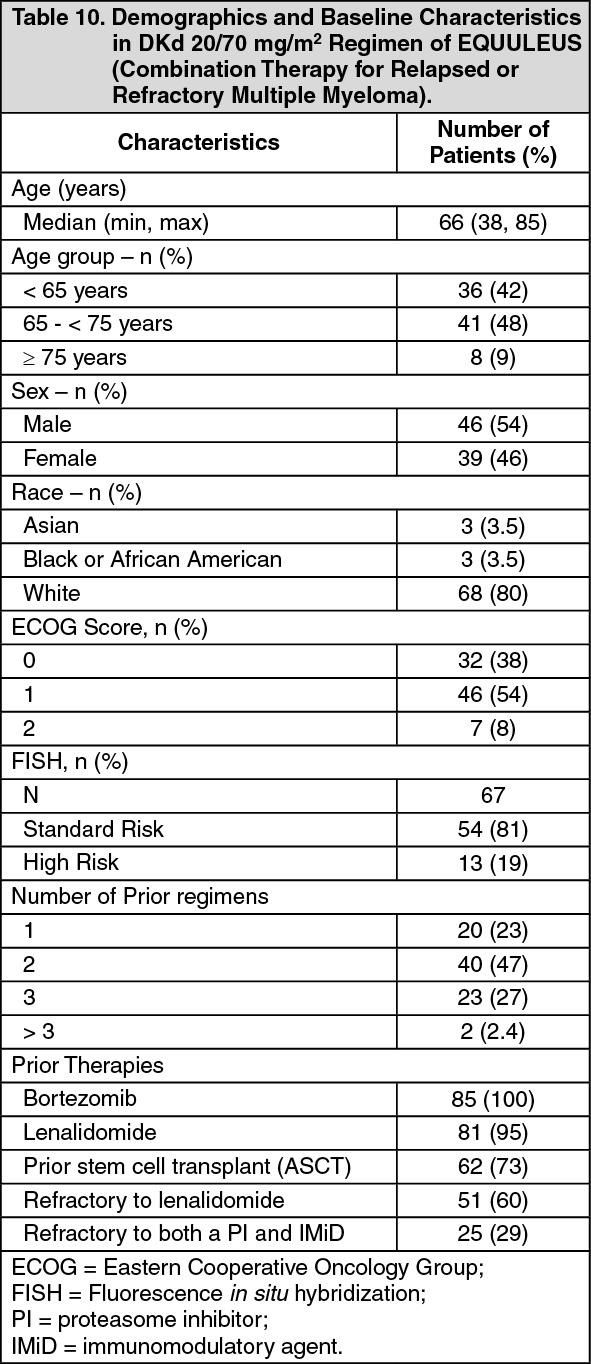 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image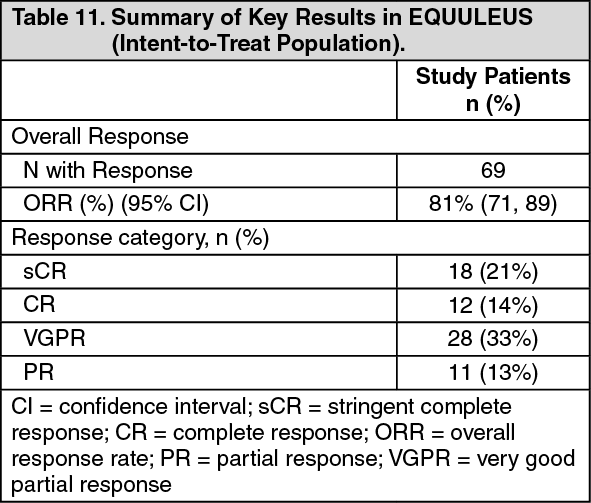 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image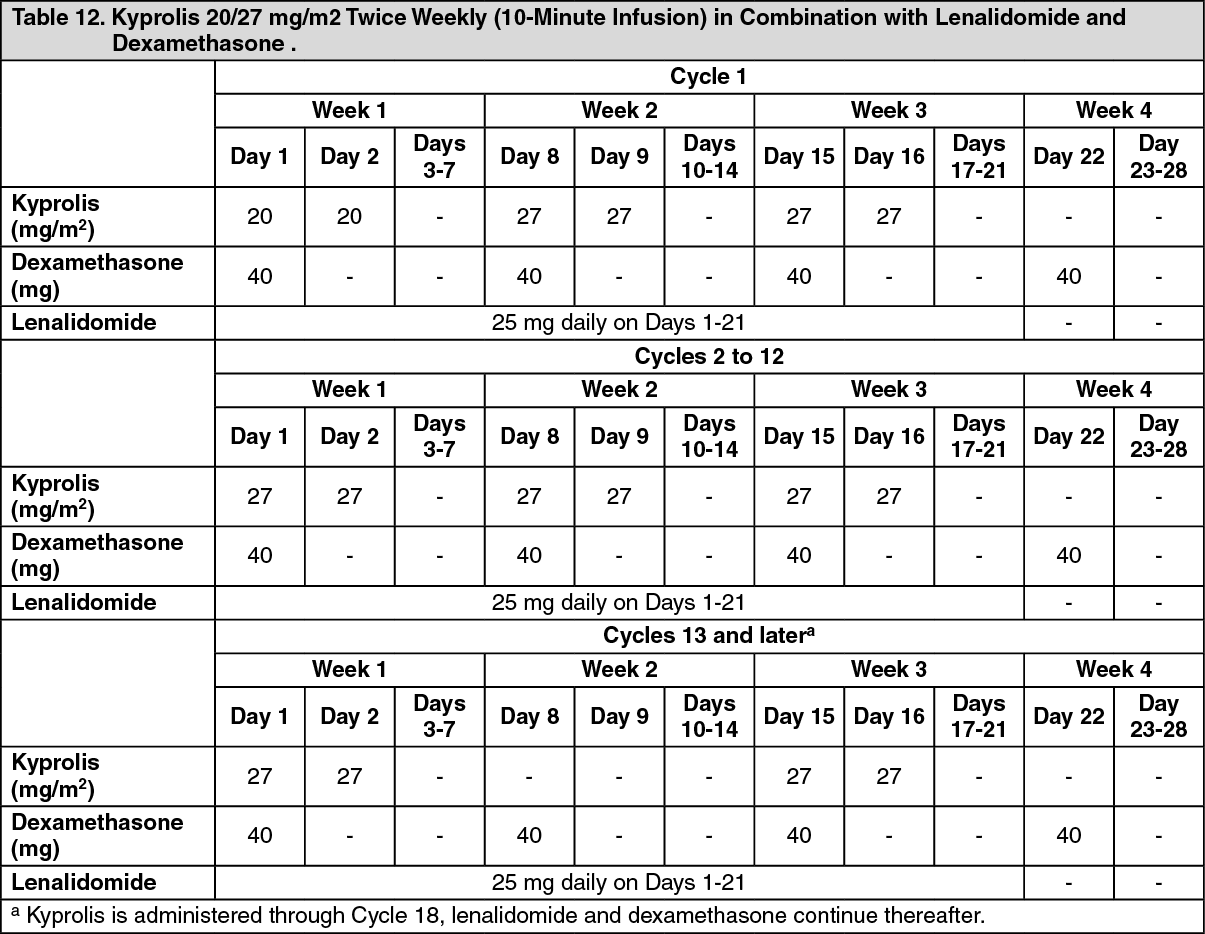 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image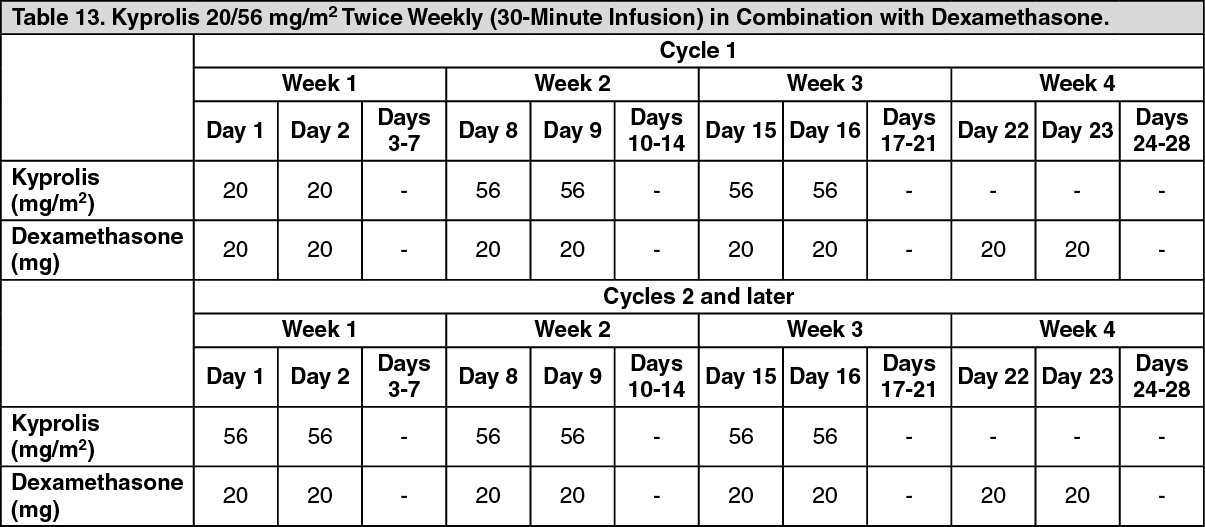 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image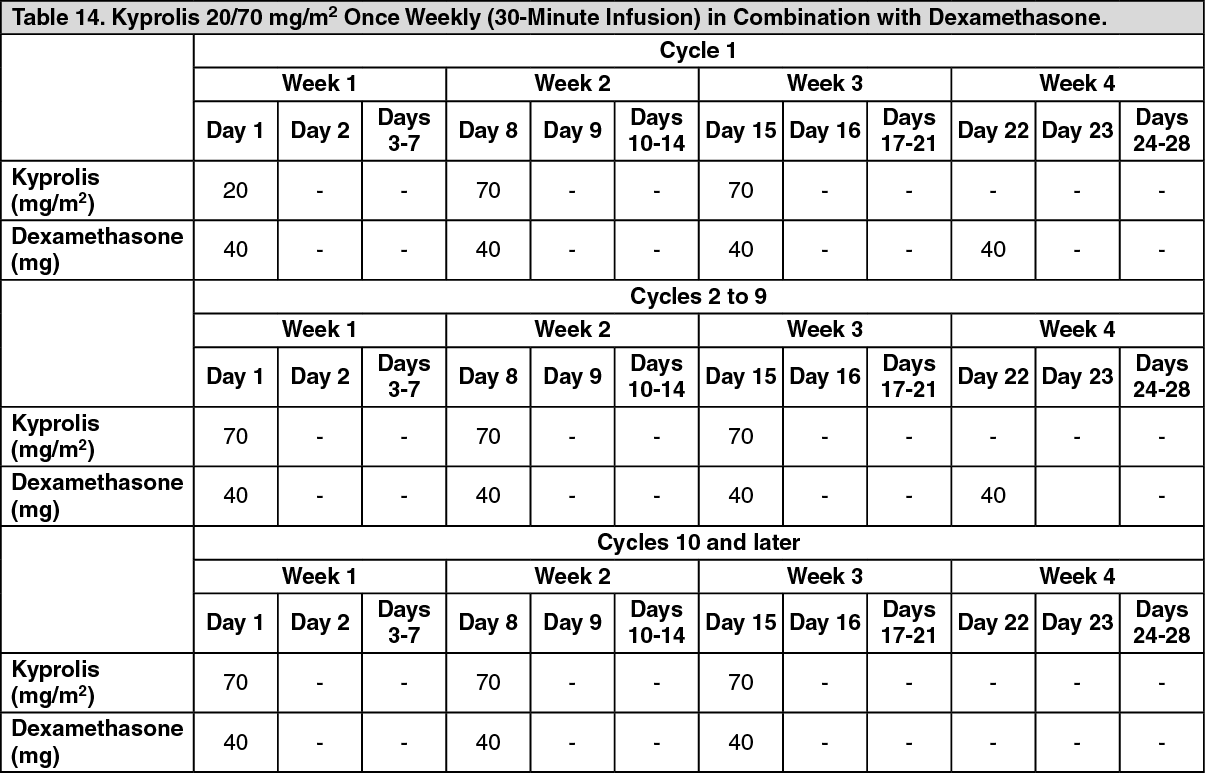 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image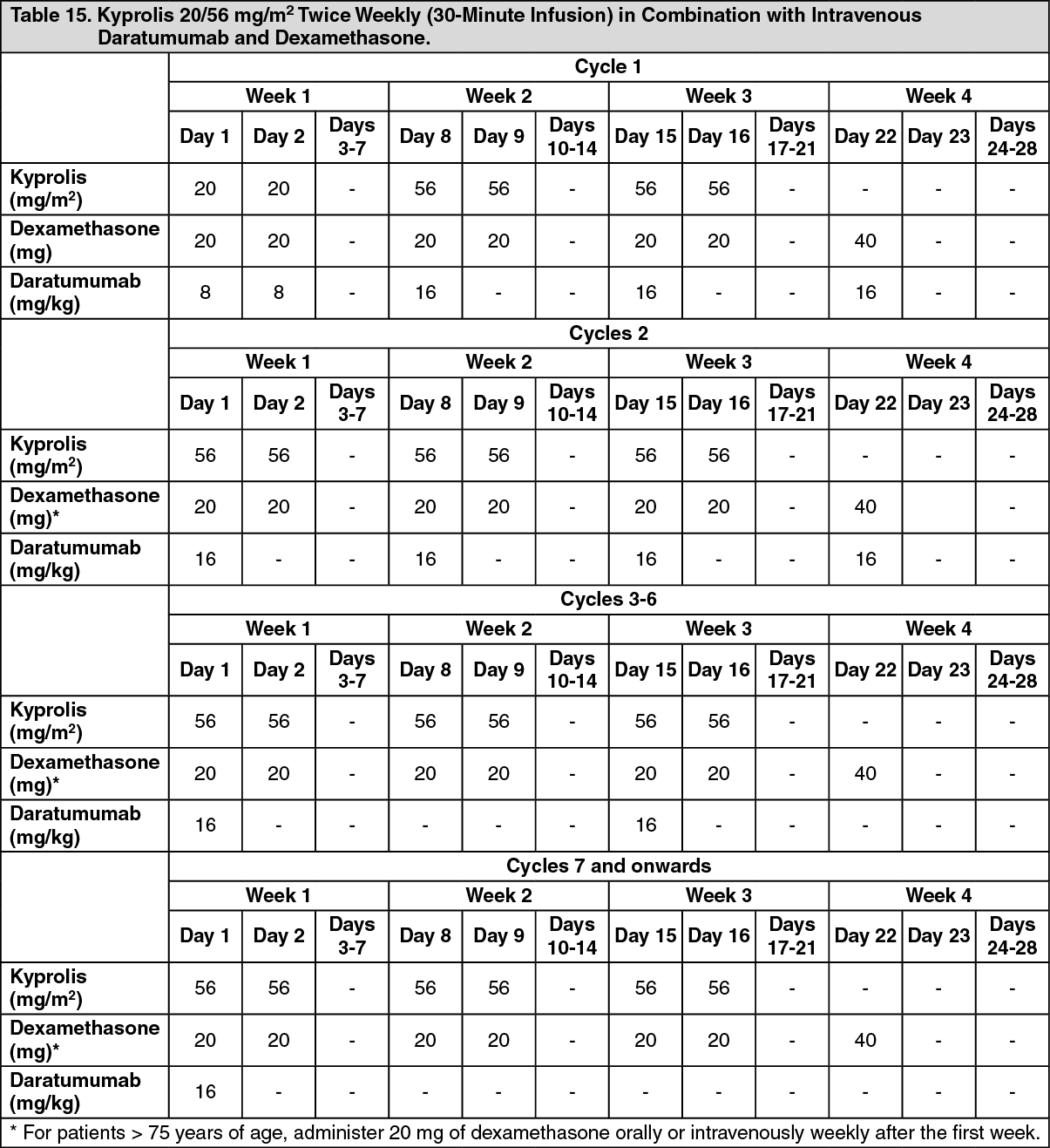 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image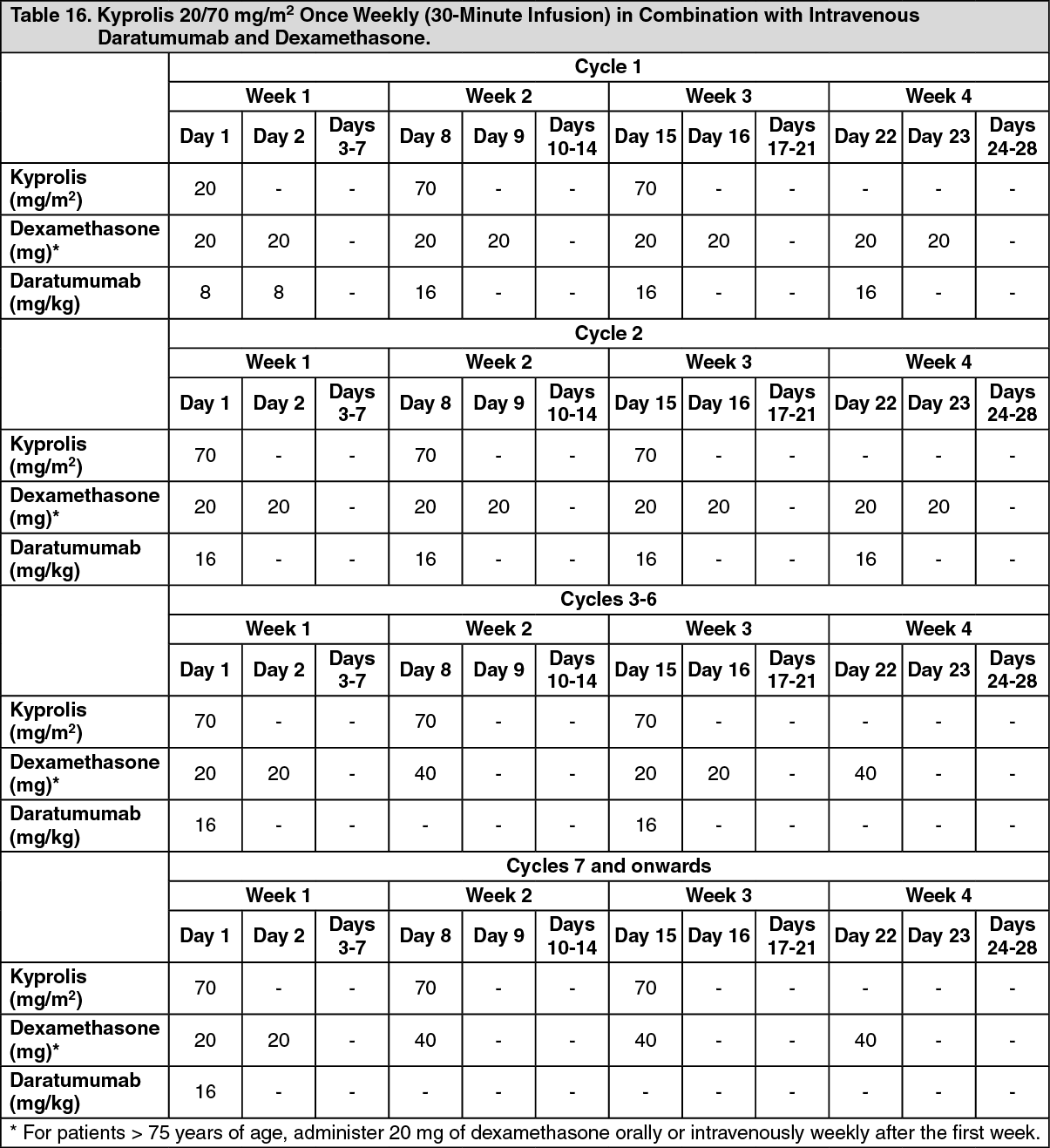 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image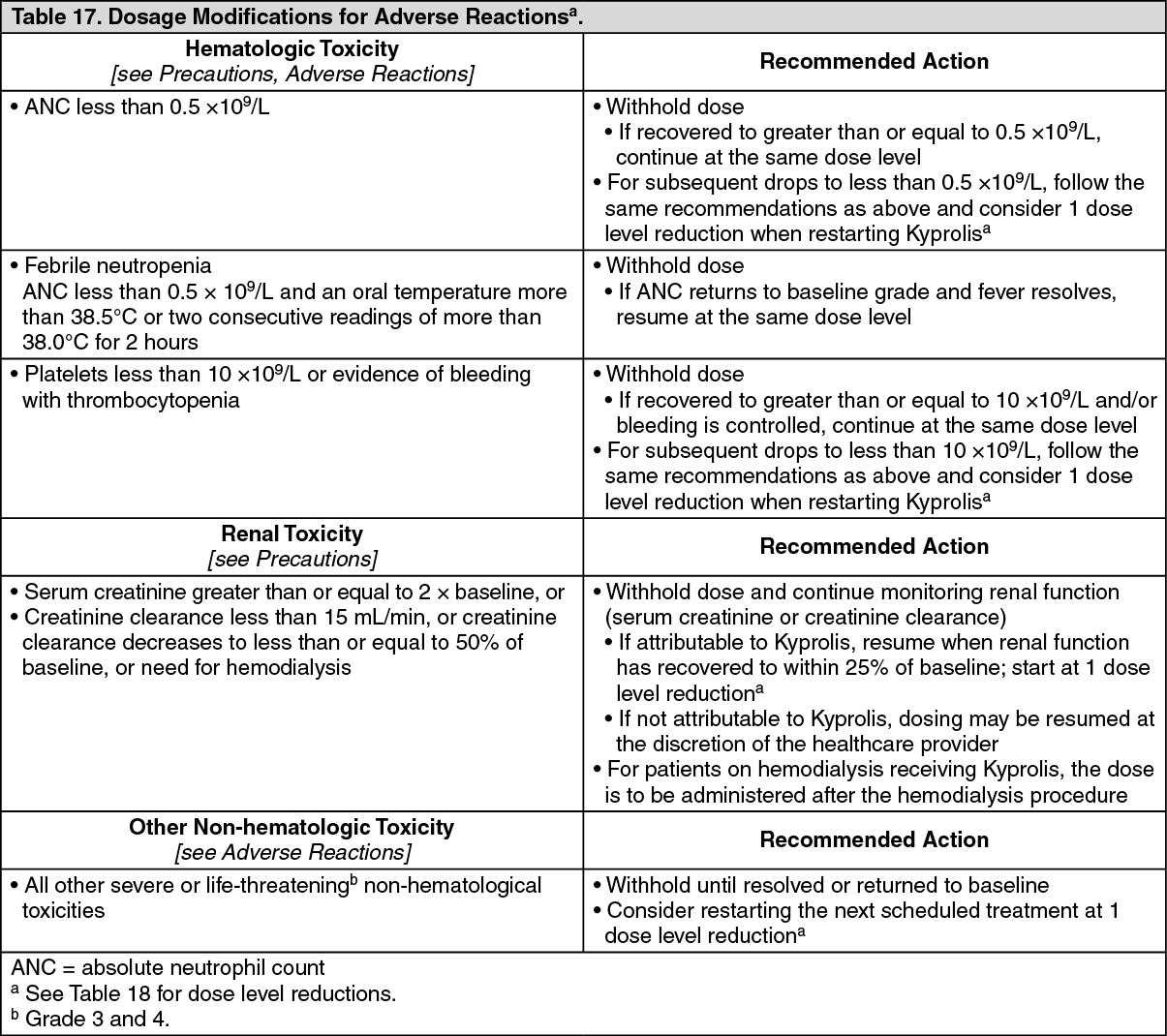 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image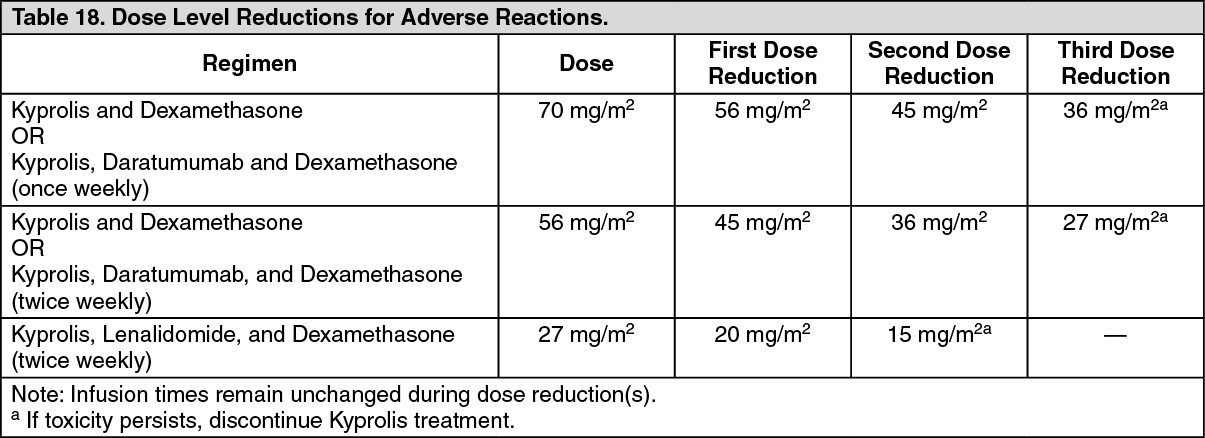 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image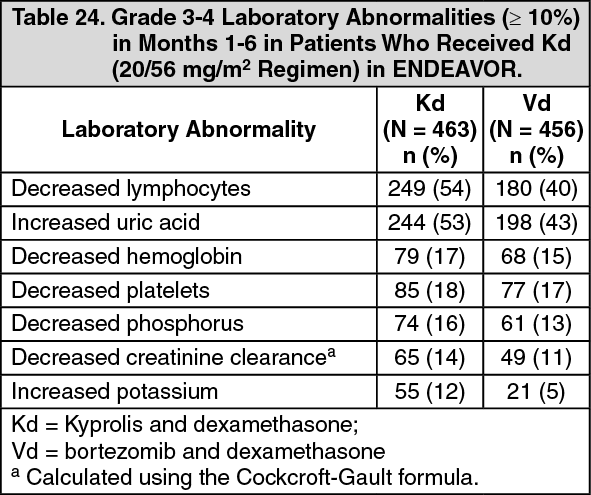 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
