


 Sign Out
Sign Out

Pharmacology: Pharmacodynamics: Mechanism of action: Abacavir and lamivudine are NRTIs, and are potent selective inhibitors of HIV-1 and HIV-2 (LAV2 and EHO) replication. Both abacavir and lamivudine are metabolised sequentially by intracellular kinases to the respective 5'-triphosphate (TP) which are the active moieties. Lamivudine-TP and carbovir-TP (the active triphosphate form of abacavir) are substrates for and competitive inhibitors of HIV reverse transcriptase (RT). However, their main antiviral activity is through incorporation of the monophosphate form into the viral DNA chain, resulting in chain termination. Abacavir and lamivudine triphosphates show significantly less affinity for host cell DNA polymerases.
No antagonistic effects in vitro were seen with lamivudine and other antiretrovirals (tested agents: didanosine, nevirapine and zidovudine). The antiviral activity of abacavir in cell culture was not antagonized when combined with the nucleoside reverse transcriptase inhibitors (NRTIs) didanosine, emtricitabine, stavudine, tenofovir or zidovudine, the non-nucleoside reverse transcriptase inhibitor (NNRTI) nevirapine, or the protease inhibitor (PI) amprenavir.
Antiviral Activity in vitro: Both abacavir and lamivudine have been shown to inhibit replication of laboratory strains and clinical isolates of HIV in a number of cell types, including transformed T cell lines, monocyte/macrophage derived lines and primary cultures of activated peripheral blood lymphocytes (PBLs) and monocyte/macrophages. The concentration of drug necessary to effect viral replication by 50% (EC50) or 50% inhibitory concentration (IC50) varied according to virus and host cell type.
The mean EC50 for abacavir against laboratory strains of HIV-1IIIB and HIV-1HXB2 ranged from 1.4 to 5.8 μM. The median or mean EC50 values for lamivudine against laboratory strains of HIV-1 ranged from 0.007 to 2.3 μM. The mean EC50 against laboratory strains of HIV-2 (LAV2 and EHO) ranged from 1.57 to 7.5 μM for abacavir and from 0.16 to 0.51 μM for lamivudine.
The EC50 values of abacavir against HIV-1 Group M subtypes (A-G) ranged from 0.002 to 1.179 μM, against Group O from 0.022 to 1.21 μM, and against HIV-2 isolates, from 0.024 to 0.49 μM. For lamivudine, the EC50 values against HIV-1 subtypes (A-G) ranged from 0.001 to 0.170 μM, against Group O from 0.030 to 0.160 μM and against HIV-2 isolates from 0.002 to 0.120 μM in peripheral blood mononuclear cells.
Baseline HIV-1 samples from therapy-naive subjects with no amino acid substitutions associated with resistance have been evaluated using either the multi-cycle Virco Antivirogram assay (n=92 from COL40263) or the the single cycle Monogram Biosciences PhenoSense assay (n=138 from ESS30009). These resulted in median EC50 values of 0.912 μM (range: 0.493 to 5.017 μM) and 1.26 μM (range 0.72 to 1.91 μM) respectively for abacavir, and median EC50 values of 0.429 μM (range: 0.200 to 2.007 μM) and 2.38 μM (1.37 to 3.68 μM) respectively for lamivudine.
Phenotypic susceptibility analyses of clinical isolates from antiretroviral-naive patients with HIV-1 Group M non-B subtypes in three studies have each reported that all viruses were fully susceptible to both abacavir and lamivudine; one study of 104 isolates that included subtypes A and A1 (n=26), C (n=1), D (n=66), and the circulating recombinant forms (CRFs) AD (n=9), CD (n=1), and a complex inter-subtype recombinant_cpx (n=1), a second study of 18 isolates including subtype G (n=14) and CRF_AG (n=4) from Nigeria, and a third study of six isolates (n=4 CRF_AG, n=1 A and n=1 undetermined) from Abidjan (Cote d'Ivoire).
HIV-1 isolates (CRF01_AE, n=12; CRF02_AG, n=12; and Subtype C or CRF_AC, n=13) from 37 untreated patients in Africa and Asia were susceptible to abacavir (IC50 fold changes <2.5), and lamivudine (IC50 fold changes<3.0), except for two CRF02_AG isolates with fold-changes of 2.9 and 3.4 for abacavir. Group O isolates from antiviral naive patients tested for lamivudine activity were highly sensitive.
The combination of abacavir and lamivudine has demonstrated antiviral activity in cell culture against non-subtype B isolates and HIV-2 isolates with equivalent antiviral activity as for subtype B isolates.
Resistance: In vivo resistance: Abacavir -resistant isolates of HIV-1 have been selected in vitro in wild-type strain HIV-1 (HXB2) and are associated with specific genotypic changes in the RT codon region (codons M184V, K65R, L74V and Y115F). Selection for the M184V mutation occurred first and resulted in a two fold increase in IC50. Continued passage in increasing concentrations of drug resulted in selection for double RT mutants 65R/184V and 74V/184V or triple RT mutant 74V/115Y/184V. Two mutations conferred a 7- to 8-fold change in abacavir susceptibility and combinations of three mutations were required to confer more than an 8-fold change in susceptibility. Passage with a zidovudine resistant clinical isolate RTMC also selected for the 184V mutation.
HIV-1 resistance to lamivudine involves the development of a M184I or, more commonly, M184V amino acid change close to the active site of the viral RT. Passage of HIV-1 (HXB2) in the presence of increasing 3TC concentrations results in high-level (>100 to >500-fold) lamivudine-resistant viruses and the RT M184I or V mutation is rapidly selected. The IC50 for wild-type HXB2 is 0.24 to 0.6 μM, while the IC50 for M184V containing HXB2 is >100 to 500 μM.
Antiviral therapy According to Genotypic/Phenotypic Resistance: In vivo resistance (Therapy-naive patients): The M184V or M184I variants arise in HIV-1 infected patients treated with lamivudine-containing antiretroviral therapy.
Isolates from most patients experiencing virological failure with a regimen containing abacavir in pivotal clinical trials showed either no NRTI-related changes from baseline (45%) or only M184V or M184I selection (45%). The overall selection frequency for M184V or M184I was high (54%), and less common was the selection of L74V (5%), K65R (1%) and Y115F (1%) (see table as follows). The inclusion of zidovudine in the regimen has been found to reduce the frequency of L74V and K65R selection in the presence of abacavir (with zidovudine: 0/40, without zidovudine: 15/192, 8%). (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageTAMs might be selected when thymidine analogs are associated with abacavir. In a meta-analysis of six clinical trials, TAMs were not selected by regimens containing abacavir without zidovudine (0/127), but were selected by regimens containing abacavir and the thymidine analogue zidovudine (22/86, 26%).
In vivo resistance (Therapy experienced patients): The M184V or M184I variants arise in HIV-1 infected patients treated with lamivudine-containing antiretroviral therapy and confer high-level resistance to lamivudine. In vitro data tend to suggest that the continuation of lamivudine in anti-retroviral regimen despite the development of M184V might provide residual anti-retroviral activity (likely through impaired viral fitness). The clinical relevance of these findings is not established. Indeed, the available clinical data are very limited and preclude any reliable conclusion in the field. In any case, initiation of susceptible NRTIs should always be preferred to maintenance of lamivudine therapy. Therefore, maintaining lamivudine therapy despite emergence of M184V mutation should only be considered in cases where no other active NRTIs are available.
Clinically significant reduction of susceptibility to abacavir has been demonstrated in clinical isolates of patients with uncontrolled viral replication, who have been pre-treated with and are resistant to other nucleoside inhibitors. In a meta-analysis of five clinical trials where ABC was added to intensify therapy, of 166 subjects, 123 (74%) had M184V/I, 50 (30%) had T215Y/F, 45 (27%) had M41L, 30 (18%) had K70R and 25 (15%) had D67N. K65R was absent and L74V and Y115F were uncommon (≤3%). Logistic regression modelling of the predictive value for genotype (adjusted for baseline plasma HIV-1RNA [vRNA], CD4+ cell count, number and duration of prior antiretroviral therapies) showed that the presence of 3 or more NRTI resistance-associated mutations was associated with reduced response at Week 4 (p=0.015) or 4 or more mutations at median Week 24 (p≤0.012). In addition, the 69 insertion complex or the Q151M mutation, usually found in combination with A62V, V75I, F77L and F116Y, cause a high level of resistance to abacavir. (See Table 2.)
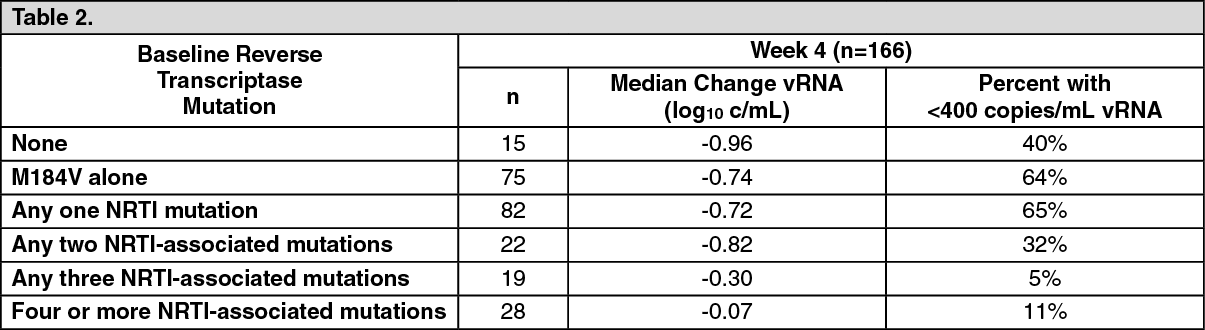 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePhenotypic resistance and cross-resistance: Phenotypic resistance to abacavir requires M184V with at least one other abacavir-selected mutation, or M184V with multiple TAMs. Phenotypic cross-resistance to other NRTIs with M184V or M184I mutation alone is limited. Zidovudine, didanosine, stavudine and tenofovir maintain their antiretroviral activities against such HIV-1 variants. The presence of M184V with K65R does give rise to cross-resistance between abacavir, tenofovir, didanosine and lamivudine, and M184V with L74V gives rise to cross-resistance between abacavir, didanosine and lamivudine. The presence of M184V with Y115F gives rise to cross-resistance between abacavir and lamivudine. Readily available genotypic drug resistance interpretation algorithms and commercially available susceptibility tests have established clinical cut offs for reduced activity for abacavir and lamivudine as separate drug entities that predict susceptibility, partial susceptibility or resistance based upon either direct measurement of susceptibility or by calculation of the HIV-1 resistance phenotype from the viral genotype. Appropriate use of abacavir and lamivudine can be guided using these currently recommended resistance algorithms.
Cross-resistance between abacavir or lamivudine and antiretrovirals from other classes e.g. PIs or NNRTIs is unlikely.
Clinical experience: Clinical experience with the combination of abacavir and lamivudine as a once daily regimen is mainly based on four studies in treatment-naive subjects, CNA30021, EPZ104057 (HEAT study), ACTG5202, and CNA109586 (ASSERT study) and two studies in treatment-experienced subjects, CAL30001 and ESS30008.
Therapy-naive patients: The combination of abacavir and lamivudine as a once daily regimen is supported by a 48 weeks multi-centre, double-blind, controlled study (CNA30021) of 770 HIV-infected, therapy-naive adults. These were primarily asymptomatic HIV infected patients (CDC stage A). They were randomised to receive either abacavir (ABC) 600 mg once daily or 300 mg twice daily, in combination with lamivudine 300 mg once daily and efavirenz 600 mg once daily. The results are summarised by subgroup in the table as follows: (See Table 3.)
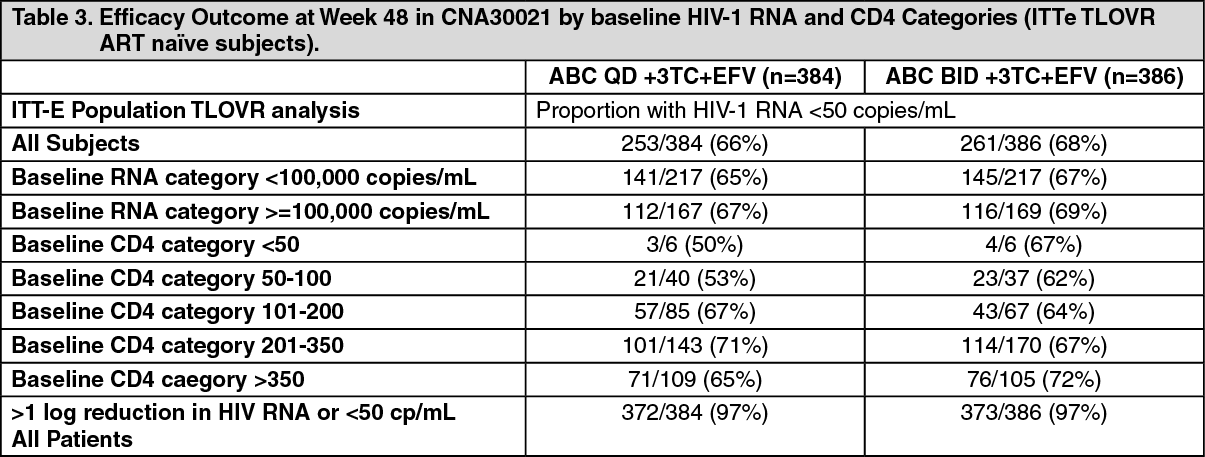 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSimilar clinical success (point estimate for treatment difference: -1.7, 95% CI -8.4, 4.9) was observed for both regimens. From these results, it can be concluded with 95% confidence that the true difference is no greater than 8.4% in favour of the twice daily regimen. This potential difference is sufficiently small to draw an overall conclusion of non-inferiority of abacavir once daily over abacavir twice daily.
There was a low, similar overall incidence of virologic failure (viral load > 50 copies/ml) in both the once and twice daily treatment groups (10% and 8% respectively). In the small sample size for genotypic analysis, there was a trend toward a higher rate of NRTI-associated mutations in the once daily versus the twice daily abacavir regimens. No firm conclusion could be drawn due to the limited data derived from this study.
There are conflicting data in some comparative studies with Kivexa i.e. HEAT, ACTG5202 and ASSERT: EPZ104057 (HEAT study) was a randomised, double-blind, placebo-matched, 96 week, multi-centre study with the primary objective of evaluating the relative efficacy of abacavir/lamivudine (ABC/3TC, 600mg/300mg) and tenofovir /emtricitabine (TDF/FTC, 300mg/200mg), each given once-daily in combination with lopinavir/ritonavir (LPV/r, 800mg/200mg) in HIV-infected, therapy-naive adults. The primary efficacy analysis was performed at week 48 with study continuation to week 96 and demonstrated non-inferiority. The results are summarised as follows: (See Table 4.)
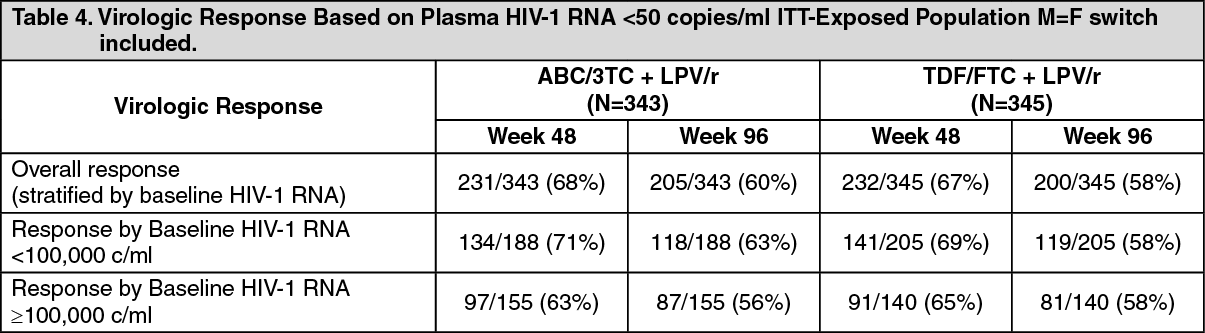 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageA similar virologic response was observed for both regimens (point estimate for treatment difference at week 48: 0.39%, 95% CI: -6.63, 7.40).
ACTG 5202 study was a multi-centre, comparative, randomised study of double-blind abacavir/lamivudine or emtricitabine/tenofovir in combination with open-label efavirenz or atazanavir/ritonavir in treatment-naive HIV-1 infected patients. Patients were stratified at screening based on plasma HIV-1 RNA levels < 100,000 and ≥ 100,000 copies/mL.
An interim analysis from ACTG 5202 revealed that abacavir/lamivudine was associated with a statistically significantly higher risk of virological failure as compared to emtricitabine/tenofovir (defined as viral load >1000 copies/mL at or after 16 weeks and before 24 weeks or HIV-RNA level >200 copies/mL at or after 24 weeks) in subjects with a screening viral load ≥100,000 copies/mL (estimated hazard ratio: 2.33, 95% CI: 1.46, 3.72, p=0.0003). The Data Safety Monitoring Board (DSMB) recommended that consideration be given to change in the therapeutic management of all subjects in the high viral load stratum due to the efficacy differences observed. The subjects in the low viral load stratum remained blinded and on-study.
Analysis of the data from subjects in the low viral load stratum showed no demonstrable difference between the nucleoside backbones in the proportion of patients free of virological failure at week 96. The results are presented as follows: 88.3% with ABC/3TC vs 90.3% with TDF/FTC when taken with atazanavir/ritonovir as third drug, treatment difference -2.0% (95% CI -7.5%, 3.4%); 87.4% with ABC/3TC vs 89.2% with TDF/FTC, when taken with efavirenz as third drug, treatment difference -1.8% (95% CI -7.5%, 3.9%).
CNA109586 (ASSERT study), a multi-centre, open label, randomised study of abacavir/lamivudine (ABC/3TC, 600mg/300mg) and tenofovir/emtricitabine (TDF/FTC, 300mg/200mg), each given once daily with efavirenz (EFV, 600mg) in ART naive, HLA-B*5701 negative, HIV-1 infected adults. The virologic results are summarised in the table as follows: (See Table 5.)
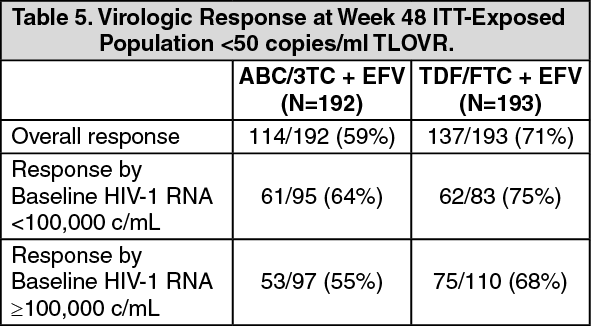 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAt week 48, a lower rate of virologic response was observed for ABC/3TC compared to TDF/FTC (point estimate for the treatment difference: 11.6%, 95% CI: 2.2, 21.1).
Therapy-experienced patients: Data from two studies, CAL30001 and ESS30008 demonstrated that Kivexa once daily has similar virological efficacy to abacavir 300 mg twice daily plus lamivudine 300 mg once daily or 150 mg twice daily in therapy-experienced patients.
In study CAL30001, 182 treatment-experienced patients with virologic failure were randomised and received treatment with either Kivexa once daily or abacavir 300 mg twice daily plus lamivudine 300 mg once daily, both in combination with tenofovir and a PI or an NNRTI for 48 weeks. Similar reductions in HIV-1 RNA as measured by average area under the curve minus baseline were observed, indicating that the Kivexa group was non-inferior to the abacavir plus lamivudine twice daily group (AAUCMB, -1.65 log10 copies/ml versus -1.83 log10 copies/ml respectively, 95% CI -0.13, 0.38). Proportions with HIV-1 RNA < 50 copies/ml (50% versus 47%) and < 400 copies/ml (54% versus 57%) at week 48 were also similar in each group (ITT population). However, as there were only moderately experienced patients included in this study with an imbalance in baseline viral load between the arms, these results should be interpreted with caution.
In study ESS30008, 260 patients with virologic suppression on a first line therapy regimen containing abacavir 300 mg plus lamivudine 150 mg, both given twice daily and a PI or NNRTI, were randomised to continue this regimen or switch to Kivexa plus a PI or NNRTI for 48 weeks. Results at 48 weeks indicated that the Kivexa group was associated with a similar virologic outcome (non-inferior) compared to the abacavir plus lamivudine group, based on proportions of subjects with HIV-1 RNA < 50 copies/ml (90% and 85% respectively, 95% CI -2.7, 13.5).
A genotypic sensitivity score (GSS) has not been established by the MAH for the abacavir/lamivudine combination. The proportion of treatment-experienced patients in the CAL30001 study with HIV-RNA <50 copies/mL at Week 48 by genotypic sensitivity score in optimised background therapy (OBT) are tabulated. The impact of major IAS-USA defined mutations to abacavir or lamivudine and multi-NRTI resistance associated mutations to the number of baseline mutations on response was also evaluated. The GSS was obtained from the Monogram reports with susceptible virus ascribed the values '1-4' based upon the numbers of drugs in the regimen and with virus with reduced susceptibility ascribed the value '0'. Genotypic sensitivity scores were not obtained for all patients at baseline. Similar proportions of patients in the once-daily and twice-daily abacavir arms of CAL30001 had GSS scores of <2 or ≥2 and successfully suppressed to <50 copies/mL by Week 48. (See Table 6.)
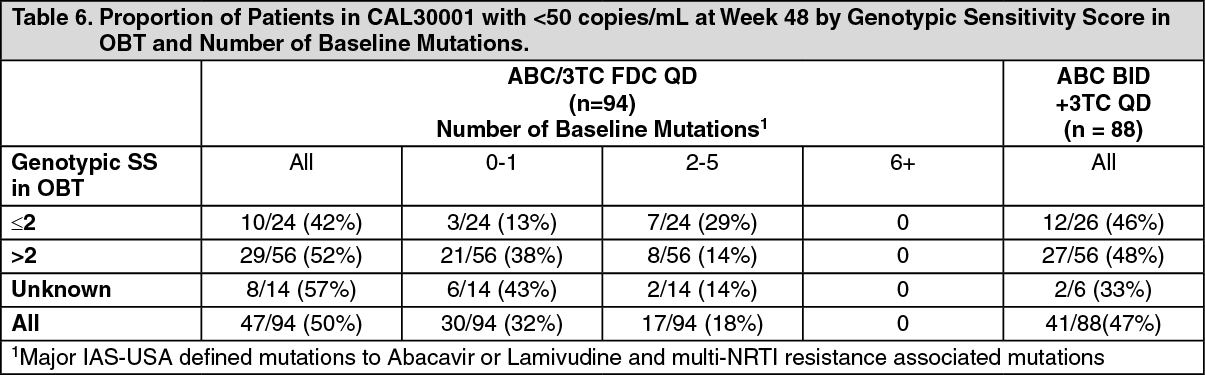 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageFor the CNA109586 (ASSERT) and CNA30021 studies in treatment-naive patients, genotype data was obtained for only a subset of patients at screening or at baseline, as well as for those patients who met virologic failure criteria. The partial patient subset of data available for CNA30021 is tabulated as follows, but must be interpreted with caution. Drug susceptibility scores were assigned for each patient's viral genotype utilising the ANRS 2009 HIV-1 genotypic drug resistance algorithm. Each susceptible drug in the regimen received a score of 1 and drugs for which the ANRS algorithm predicts resistance were ascribed the value '0'. (See Table 7.)
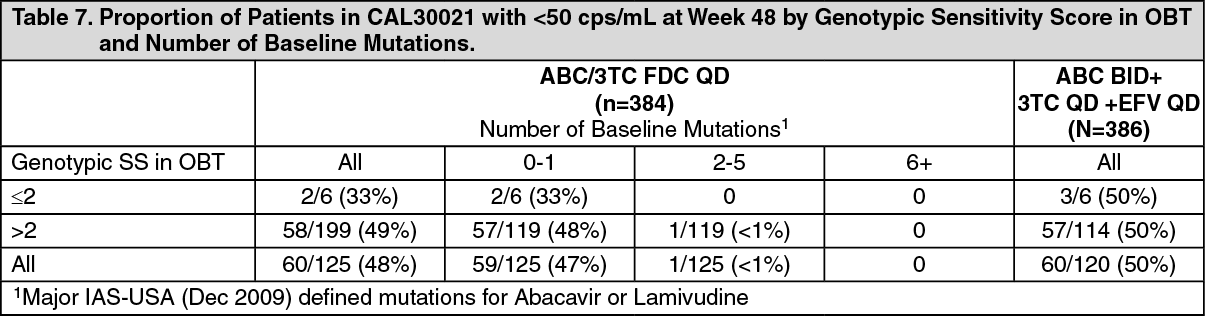 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePaediatric population: A comparison of a regimen including once daily versus twice daily dosing of abacavir and lamivudine was undertaken within a randomised, multicentre, controlled study of HIV-infected, paediatric patients. 1206 paediatric patients aged 3 months to 17 years enrolled in the ARROW Trial (COL105677) and were dosed according to the weight - band dosing recommendations in the World Health Organisation treatment guidelines (Antiretroviral therapy of HIV infection in infants and children, 2006). After 36 weeks on a regimen including twice daily abacavir and lamivudine, 669 eligible subjects were randomised to either continue twice daily dosing or switch to once daily abacavir and lamivudine for at least an additional 96 weeks. Within this population, 104 patients, weighing at least 25 kg, received 600 mg abacavir and 300 mg lamivudine as Kivexa once daily, with a median duration of exposure of 596 days.
Among the 669 subjects randomized in this study (from 12 months to ≤17 years old), the abacavir/lamivudine once daily dosing group was demonstrated to be non-inferior to the twice daily group according to the pre-specified non-inferiority margin of -12%, for the primary endpoint of <80 c/mL at Week 48 as well as at Week 96 (secondary endpoint) and all other thresholds tested (<200c/mL, <400c/mL, <1000c/mL), which all fell well within this non-inferiority margin. Subgroup analyses testing for heterogeneity of once versus twice daily demonstrated no significant effect of sex, age, or viral load at randomisation. Conclusions supported non-inferiority regardless of analysis method.
Among the 104 patients who received Kivexa, including the ones who were between 40 kg and 25 kg, the viral suppression was similar.
Pharmacokinetics: The fixed-dose combination tablet of abacavir/lamivudine (FDC) has been shown to be bioequivalent to lamivudine and abacavir administered separately. This was demonstrated in a single dose, 3-way crossover bioequivalence study of FDC (fasted) versus 2 x 300 mg abacavir tablets plus 2 x 150 mg lamivudine tablets (fasted) versus FDC administered with a high fat meal, in healthy volunteers (n = 30). In the fasted state there was no significant difference in the extent of absorption, as measured by the area under the plasma concentration-time curve (AUC) and maximal peak concentration (Cmax), of each component. There was also no clinically significant food effect observed between administration of FDC in the fasted or fed state. These results indicate that FDC can be taken with or without food. The pharmacokinetic properties of lamivudine and abacavir are described as follows.
Absorption: Abacavir and lamivudine are rapidly and well absorbed from the gastro-intestinal tract following oral administration. The absolute bioavailability of oral abacavir and lamivudine in adults is about 83% and 80-85% respectively. The mean time to maximal serum concentrations (tmax) is about 1.5 hours and 1.0 hour for abacavir and lamivudine, respectively. Following a single dose of 600 mg of abacavir, the mean (CV) Cmax is 4.26 μg/ml (28%) and the mean (CV) AUC∞ is 11.95 μg.h/ml (21%). Following multiple-dose oral administration of lamivudine 300 mg once daily for seven days, the mean (CV) steady-state Cmax is 2.04 μg/ml (26%) and the mean (CV) AUC24 is 8.87 μg.h/ml (21%).
Distribution: Intravenous studies with abacavir and lamivudine showed that the mean apparent volume of distribution is 0.8 and 1.3 l/kg respectively. Plasma protein binding studies in vitro indicate that abacavir binds only low to moderately (~49%) to human plasma proteins at therapeutic concentrations. Lamivudine exhibits linear pharmacokinetics over the therapeutic dose range and displays limited plasma protein binding in vitro (< 36%). This indicates a low likelihood for interactions with other medicinal products through plasma protein binding displacement.
Data show that abacavir and lamivudine penetrate the central nervous system (CNS) and reach the cerebrospinal fluid (CSF). Studies with abacavir demonstrate a CSF to plasma AUC ratio of between 30 to 44%. The observed values of the peak concentrations are 9 fold greater than the IC50 of abacavir of 0.08 μg/ml or 0.26 μM when abacavir is given at 600 mg twice daily. The mean ratio of CSF/serum lamivudine concentrations 2-4 hours after oral administration was approximately 12%. The true extent of CNS penetration of lamivudine and its relationship with any clinical efficacy is unknown.
Biotransformation: Abacavir is primarily metabolised by the liver with approximately 2% of the administered dose being renally excreted, as unchanged compound. The primary pathways of metabolism in man are by alcohol dehydrogenase and by glucuronidation to produce the 5'-carboxylic acid and 5'-glucuronide which account for about 66% of the administered dose. These metabolites are excreted in the urine.
Metabolism of lamivudine is a minor route of elimination. Lamivudine is predominately cleared by renal excretion of unchanged lamivudine. The likelihood of metabolic drug interactions with lamivudine is low due to the small extent of hepatic metabolism (5-10%).
Elimination: The mean half-life of abacavir is about 1.5 hours. Following multiple oral doses of abacavir 300 mg twice a day there is no significant accumulation of abacavir. Elimination of abacavir is via hepatic metabolism with subsequent excretion of metabolites primarily in the urine. The metabolites and unchanged abacavir account for about 83% of the administered abacavir dose in the urine. The remainder is eliminated in the faeces.
The observed lamivudine half-life of elimination is 5 to 7 hours. The mean systemic clearance of lamivudine is approximately 0.32 l/h/kg, predominantly by renal clearance (> 70%) via the organic cationic transport system. Studies in patients with renal impairment show lamivudine elimination is affected by renal dysfunction. Kivexa is not recommended for use in patients with a creatinine clearance < 50 ml/min as necessary dose adjustment cannot be made (see Dosage & Administration).
Intracellular pharmacokinetics: In a study of 20 HIV-infected patients receiving abacavir 300 mg twice daily, with only one 300 mg dose taken prior to the 24 hour sampling period, the geometric mean terminal carbovir-TP intracellular half-life at steady-state was 20.6 hours, compared to the geometric mean abacavir plasma half-life in this study of 2.6 hours. In a crossover study in 27 HIV-infected patients, intracellular carbovir-TP exposures were higher for the abacavir 600 mg once daily regimen (AUC24,ss + 32 %, Cmax24,ss + 99 % and Ctrough + 18 %) compared to the 300 mg twice daily regimen. For patients receiving lamivudine 300 mg once daily, the terminal intracellular half-life of lamivudine-TP was prolonged to 16-19 hours, compared to the plasma lamivudine half-life of 5-7 hours. In a crossover study in 60 healthy volunteers, intracellular lamivudine-TP pharmacokinetic parameters were similar (AUC24,ss and Cmax24,ss) or lower (Ctrough - 24 %) for the lamivudine 300 mg once daily regimen compared to the lamivudine 150 mg twice daily regimen. Overall, these data support the use of lamivudine 300 mg and abacavir 600 mg once daily for the treatment of HIV-infected patients. Additionally, the efficacy and safety of this combination given once daily has been demonstrated in a pivotal clinical study (CNA30021- see Clinical experience as previously mentioned).
Special patient populations: Hepatic impairment: Pharmacokinetic data has been obtained for abacavir and lamivudine separately.
Abacavir is metabolised primarily by the liver. The pharmacokinetics of abacavir have been studied in patients with mild hepatic impairment (Child-Pugh score 5-6) receiving a single 600 mg dose; the median (range) AUC value was 24.1 (10.4 to 54.8) ug.h/ml. The results showed that there was a mean (90%CI) increase of 1.89 fold [1.32; 2.70] in the abacavir AUC, and 1.58 [1.22; 2.04] fold in the elimination half-life. No definitive recommendation on dosage reduction is possible in patients with mild hepatic impairment due to substantial variability of abacavir exposure.
Data obtained in patients with moderate to severe hepatic impairment show that lamivudine pharmacokinetics are not significantly affected by hepatic dysfunction.
Based on data obtained for abacavir, Kivexa is not recommended in patients with moderate or severe hepatic impairment.
Renal impairment: Pharmacokinetic data have been obtained for lamivudine and abacavir alone. Abacavir is primarily metabolised by the liver with approximately 2% of abacavir excreted unchanged in the urine. The pharmacokinetics of abacavir in patients with end-stage renal disease is similar to patients with normal renal function. Studies with lamivudine show that plasma concentrations (AUC) are increased in patients with renal dysfunction due to decreased clearance. Kivexa is not recommended for use in patients with a creatinine clearance of < 50 ml/min as necessary dose adjustment cannot be made.
Elderly: No pharmacokinetic data are available in patients over 65 years of age.
Children: Abacavir is rapidly and well absorbed from oral formulations when administered to children. Paediatric pharmacokinetic studies have demonstrated that once daily dosing provides equivalent AUC24 to twice daily dosing of the same total daily dose for both oral solution and tablet formulations.
The absolute bioavailability of lamivudine (approximately 58 to 66%) was lower and more variable in paediatric patients under 12 years of age. However, paediatric pharmacokinetic studies with tablet formulations have demonstrated that once daily dosing provides equivalent AUC24 to twice daily dosing of the same total daily dose.
Toxicology: Preclinical safety data: With the exception of a negative in vivo rat micronucleus test, there are no data available on the effects of the combination of abacavir and lamivudine in animals.
Mutagenicity and carcinogenicity: Neither abacavir nor lamivudine were mutagenic in bacterial tests, but consistent with other nucleoside analogues, they inhibit cellular DNA replication in in vitro mammalian tests such as the mouse lymphoma assay. The results of an in vivo rat micronucleus test with abacavir and lamivudine in combination were negative.
Lamivudine has not shown any genotoxic activity in the in vivo studies at doses that gave plasma concentrations up to 40-50 times higher than clinical plasma concentrations. Abacavir has a weak potential to cause chromosomal damage both in vitro and in vivo at high tested concentrations.
The carcinogenic potential of a combination of abacavir and lamivudine has not been tested. In long-term oral carcinogenicity studies in rats and mice, lamivudine did not show any carcinogenic potential. Carcinogenicity studies with orally administered abacavir in mice and rats showed an increase in the incidence of malignant and non-malignant tumours. Malignant tumours occurred in the preputial gland of males and the clitoral gland of females of both species, and in rats in the thyroid gland of males and in the liver, urinary bladder, lymph nodes and the subcutis of females.
The majority of these tumours occurred at the highest abacavir dose of 330 mg/kg/day in mice and 600 mg/kg/day in rats. The exception was the preputial gland tumour which occurred at a dose of 110 mg/kg in mice. The systemic exposure at the no effect level in mice and rats was equivalent to 3 and 7 times the human systemic exposure during therapy. While the clinical relevance of these findings is unknown, these data suggest that a carcinogenic risk to humans is outweighed by the potential clinical benefit.
Repeat-dose toxicity: In toxicology studies abacavir was shown to increase liver weights in rats and monkeys. The clinical relevance of this is unknown. There is no evidence from clinical studies that abacavir is hepatotoxic. Additionally, autoinduction of abacavir metabolism or induction of the metabolism of other medicinal products hepatically metabolised has not been observed in man.
Mild myocardial degeneration in the heart of mice and rats was observed following administration of abacavir for two years. The systemic exposures were equivalent to 7 to 24 times the expected systemic exposure in humans. The clinical relevance of this finding has not been determined.
Reproductive toxicology: In reproductive toxicity studies in animals, lamivudine and abacavir were shown to cross the placenta.
Lamivudine was not teratogenic in animal studies but there were indications of an increase in early embryonic deaths in rabbits at relatively low systemic exposures, comparable to those achieved in humans. A similar effect was not seen in rats even at very high systemic exposure.
Abacavir demonstrated toxicity to the developing embryo and foetus in rats, but not in rabbits. These findings included decreased foetal body weight, foetal oedema, and an increase in skeletal variations/malformations, early intra-uterine deaths and still births. No conclusion can be drawn with regard to the teratogenic potential of abacavir because of this embryo-foetal toxicity.
A fertility study in rats has shown that abacavir and lamivudine had no effect on male or female fertility.