


 Sign Out
Sign Out


Haematological adverse drug reactions (any Common Terminology Criteria for Adverse Events [CTCAE] grade) included anaemia (83.8%), thrombocytopenia (80.5%) and neutropenia (20.8%).
Anaemia, thrombocytopenia and neutropenia are dose-related effects.
The three most frequent non-haematological adverse drug reactions were bruising (33.3%), other bleeding (including epistaxis, post-procedural haemorrhage and haematuria) (24.3%) and dizziness (21.9%).
The three most frequent non-haematological laboratory abnormalities were increased alanine aminotransferase (40.7%), raised aspartate aminotransferase (31.5%) and hypertriglyceridaemia (25.2%). In phase 3 clinical studies in MF, neither CTCAE grade 3 or 4 hypertriglyceridaemia or increased aspartate aminotransferase, nor CTCAE grade 4 raised alanine aminotransferase or hypercholesterolaemia were observed.
Discontinuation due to adverse events, regardless of causality, was observed in 30.0% of patients.
Polycythaemia vera: Haematological adverse reactions (any CTCAE grade) included anaemia (61.8%) and thrombocytopenia (25.0%). Anaemia and thrombocytopenia CTCAE grade 3 or 4 were reported in respectively 2.9% and 2.6%.
The three most frequent non-haematological adverse reactions were weight gain (20.3%), dizziness (19.4%) and headache (17.9%).
The three most frequent non-haematological laboratory abnormalities (any CTCAE grade) identified as adverse reactions were raised alanine aminotransferase (45.3%), raised aspartate aminotransferase (42.6%), and hypercholesterolaemia (34.7%). No CTCAE grade 4 increased alanine aminotransferase or hypercholesterolaemia, and one CTCAE grade 4 raised aspartate aminotransferase were observed.
Discontinuation due to adverse events, regardless of causality, was observed in 19.4% of patients.
Acute GvHD: The most frequently reported overall adverse drug reactions were thrombocytopenia, anaemia and neutropenia.
Haematological laboratory abnormalities identified as adverse drug reactions included thrombocytopenia (85.2%), anaemia (75.0%) and neutropenia (65.1%). Grade 3 anaemia was reported in 47.7% of patients (grade 4 not applicable per CTCAE v4.03). Grade 3 or 4 thrombocytopenia were reported in 31.3% and 47.7% of patients, respectively.
The three most frequent non-haematological adverse drug reactions were cytomegalovirus (CMV) infection (32.3%), sepsis (25.4%) and urinary tract infections (17.9%).
The three most frequent non-haematological laboratory abnormalities identified as adverse drug reactions were increased alanine aminotransferase (54.9%), increased aspartate aminotransferase (52.3%) and hypercholesterolaemia (49.2%). The majority were of grade 1 and 2.
Discontinuation due to adverse events, regardless of causality, was observed in 29.4% of patients.
Chronic GvHD: The most frequently reported overall adverse drug reactions were anaemia, hypercholesterolemia and increased aspartate aminotransferase.
Haematological laboratory abnormalities identified as adverse drug reactions included anaemia (68.6%), thrombocytopenia (34.4%) and neutropenia (36.2%). Grade 3 anaemia was reported in 14.8% of patients (grade 4 not applicable per CTCAE v4.03). Grade 3 and 4 neutropenia were reported in 9.5% and 6.7% of patients, respectively.
The three most frequent non-haematological adverse drug reactions were hypertension (15.0%), headache (10.2%) and urinary tract infections (9.3%).
The three most frequent non-haematological laboratory abnormalities identified as adverse drug reactions were hypercholesterolaemia (52.3%), increased aspartate aminotransferase (52.2%) and increased alanine aminotransferase (43.1%). The majority were grade 1 and 2.
Discontinuation due to adverse events, regardless of causality, was observed in 18.1% of patients.
Tabulated list of adverse drug reactions from clinical studies: The safety of Jakavi in MF patients was evaluated using the long-term follow-up data from two phase 3 studies (COMFORT-I and COMFORT-II) including data from patients initially randomised to ruxolitinib (n=301) and patients who received ruxolitinib after crossing over from control treatments (n=156). The median exposure upon which the adverse drug reaction frequencies categories for MF patients are based was 30.5 months (range 0.3 to 68.1 months).
The safety of Jakavi in PV patients was evaluated using the long-term follow-up data from two phase 3 studies (RESPONSE, RESPONSE 2) including data from patients initially randomised to ruxolitinib (n=184) and patients who received ruxolitinib after crossing over from control treatments (n=156). The median exposure upon which the adverse drug reaction frequency categories for PV patients are based was 41.7 months (range 0.03 to 59.7 months).
The safety of Jakavi in acute GvHD patients was evaluated in the phase 3 study REACH2, including data from patients initially randomised to Jakavi (n=152) and patients who received Jakavi after crossing over from the best available therapy (BAT) arm (n=49). The median exposure upon which the adverse drug reaction frequency categories were based was 8.9 weeks (range 0.3 to 66.1 weeks).
The safety of Jakavi in chronic GvHD patients was evaluated in the phase 3 study REACH3, including data from patients initially randomised to Jakavi (n=165) and patients who received Jakavi after crossing over from BAT (n=61). The median exposure upon which the adverse drug reaction frequency categories were based was 41.4 weeks (range 0.7 to 127.3 weeks).
In the clinical study programme the severity of adverse drug reactions was assessed based on the CTCAE, defining grade 1 = mild, grade 2 = moderate, grade 3 = severe, grade 4=life-threatening or disabling, grade 5 = death.
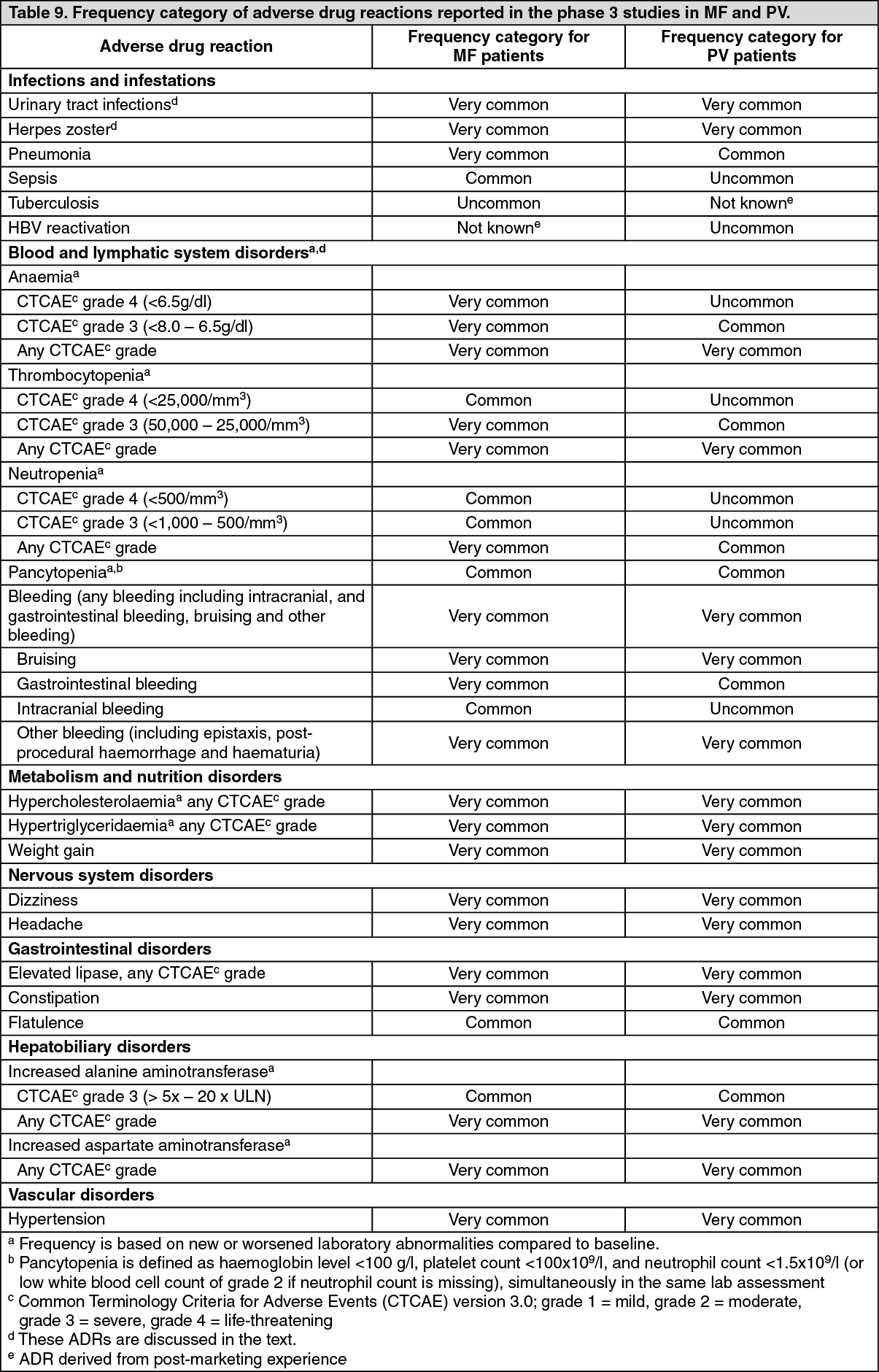
Adverse drug reactions from clinical studies in MF and PV (Table 9) and in acute and chronic GvHD are listed by MedDRA system organ class. Within each system organ class, the adverse drug reactions are ranked by frequency, with the most frequent reactions first. In addition, the corresponding frequency category for each adverse drug reaction is based on the following convention: very common (≥1/10); common (≥1/100 to <1/10); uncommon (≥1/1,000 to <1/100); rare (≥1/10,000 to <1/1,000); very rare (<1/10,000); not known (cannot be estimated from the available data). (See Table 9.)
 Click on icon to see table/diagram/image
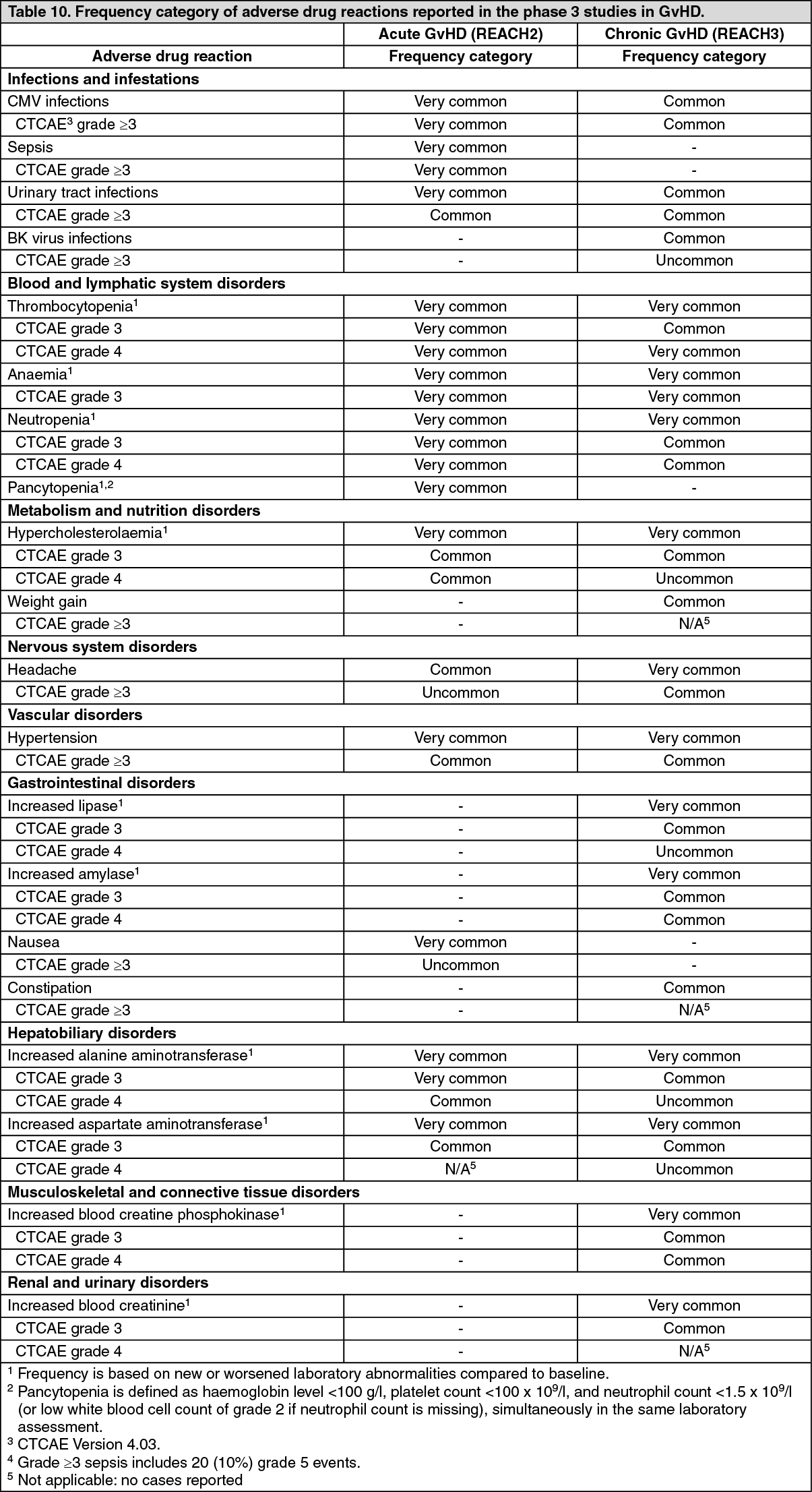
Click on icon to see table/diagram/imageUpon discontinuation, MF patients may experience a return of MF symptoms such as fatigue, bone pain, fever, pruritus, night sweats, symptomatic splenomegaly and weight loss. In clinical studies in MF the total symptom score for MF symptoms gradually returned to baseline value within 7 days after dose discontinuation (see Precautions). (See Table 10.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDescription of selected adverse drug reactions: Anaemia: In phase 3 clinical studies in MF, median time to onset of first CTCAE grade 2 or higher anaemia was 1.5 months. One patient (0.3%) discontinued treatment because of anaemia.
In patients receiving ruxolitinib mean decreases in haemoglobin reached a nadir of approximately 10 g/litre below baseline after 8 to 12 weeks of therapy and then gradually recovered to reach a new steady state that was approximately 5 g/litre below baseline. This pattern was observed in patients regardless of whether they had received transfusion during therapy.
In the randomised, placebo-controlled study COMFORT-I 60.6% of Jakavi-treated MF patients and 37.7% of placebo-treated MF patients received red blood cell transfusions during randomised treatment. In the COMFORT-II study the rate of packed red blood cell transfusions was 53.4% in the Jakavi arm and 41.1% in the best available therapy arm.
In the randomised period of the pivotal studies, anaemia was less frequent in PV patients than in MF patients (40.8% versus 82.4%). In the PV population, the CTCAE grade 3 and 4 events were reported in 2.7%, while in the MF patients the frequency was 42.56%.
In the phase 3 acute and chronic GvHD studies, anaemia CTCAE grade 3 was reported in 47.7% and 14.8% of patients, respectively.
Thrombocytopenia: In the phase 3 clinical studies in MF, in patients who developed grade 3 or 4 thrombocytopenia, the median time to onset was approximately 8 weeks. Thrombocytopenia was generally reversible with dose reduction or dose interruption. The median time to recovery of platelet counts above 50,000/mm3 was 14 days. During the randomised period, platelet transfusions were administered to 4.7% of patients receiving ruxolitinib and to 4.0% of patients receiving control regimens. Discontinuation of treatment because of thrombocytopenia occurred in 0.7% of patients receiving ruxolitinib and 0.9% of patients receiving control regimens. Patients with a platelet count of 100,000/mm3 to 200,000/mm3 before starting ruxolitinib had a higher frequency of grade 3 or 4 thrombocytopenia compared to patients with platelet count >200,000/mm3 (64.2% versus 38.5%).
In the randomised period of the pivotal studies, the rate of patients experiencing thrombocytopenia was lower in PV (16.8%) patients compared to MF (69.8%) patients. The frequency of severe (i.e. CTCAE grade 3 and 4) thrombocytopenia was lower in PV (2.7%) than in MF (11.6%) patients.
In the phase 3 acute GvHD study, grade 3 and 4 thrombocytopenia was observed in 31.3% and 47.7% of patients, respectively. In the phase 3 chronic GvHD study, grade 3 and 4 thrombocytopenia was lower (5.9% and 10.7%) than in acute GvHD.
Neutropenia: In the phase 3 clinical studies in MF, in patients who developed grade 3 or 4 neutropenia, the median time to onset was 12 weeks. During the randomised period, dose holding or reductions due to neutropenia were reported in 1.0% of patients, and 0.3% of patients discontinued treatment because of neutropenia.
In the randomised period of the phase 3 studies in PV patients, neutropenia was reported in 1.6% of patients exposed to ruxolitinib compared to 7% in reference treatments. In the ruxolitinib arm one patient developed CTCAE grade 4 neutropenia. An extended follow-up of patients treated with ruxolitinib showed 2 patients reporting CTCAE grade 4 neutropenia.
In the phase 3 acute GvHD study, grade 3 and 4 neutropenia was obseerved in 17.9% and 20.6% of patients, respectively. In these phase 3 chronic GvHD study grade 3 and 4 neutropenia was lower (9.5% and 6.7%) than in acute GvHD.
Bleeding: In the phase 3 pivotal studies in MF bleeding events (including intracranial and gastrointestinal, bruising and other bleeding events) were reported in 32.6% of patients exposed to ruxolitinib and 23.2% of patients exposed to the reference treatments (placebo or best available therapy). The frequency of grade 3-4 events was similar for patients treated with ruxolitinib or reference treatments (4.7% versus 3.1%). Most of the patients with bleeding events during the treatment reported bruising (65.3%). Bruising events were more frequently reported in patients taking ruxolitinib compared with the reference treatments (21.3% versus 11.6%). Intracranial bleeding was reported in 1% of patients exposed to ruxolitinib and 0.9% exposed to reference treatments. Gastrointestinal bleeding was reported in 5.0% of patients exposed to ruxolitinib compared to 3.1% exposed to reference treatments. Other bleeding events (including events such as epistaxis, post-procedural haemorrhage and haematuria) were reported in 13.3% of patients treated with ruxolitinib and 10.3% treated with reference treatments.
During the long-term follow-up of phase 3 clinical studies in MF, the cumulative frequency of bleeding events increased proportionally to the increase in the follow-up time. Bruising events were the most frequently reported bleeding events (33.3%). Intracranial and gastrointestinal bleeding events were reported in 1.3% and 10.1% of patients respectively.
In the comparative period of phase 3 studies in PV patients, bleeding events (including intracranial and gastrointestinal, bruising and other bleeding events) were reported in 16.8% of patients treated with ruxolitinib, 15.3% of patients receiving best available therapy in RESPONSE study and 12.0% of patients receiving best available therapy in RESPONSE 2 study. Bruising was reported in 10.3% of patients treated with ruxolitinib, 8.1% of patients receiving best available therapy in RESPONSE study and 2.7% of patients receiving best available therapy in RESPONSE 2 study. No intracranial bleeding or gastrointestinal haemorrhage events were reported in patients receiving ruxolitinib. One patient treated with ruxolitinib experienced a grade 3 bleeding event (post-procedural bleeding); no grade 4 bleeding was reported. Other bleeding events (including events such as epistaxis, post-procedural haemorrhage, gingival bleeding) were reported in 8.7% of patients treated with ruxolitinib, 6.3% of patients treated with best available therapy in RESPONSE study and 6.7% of patients treated with best available therapy in RESPONSE 2 study.
During the long-term follow-up of phase 3 studies in PV, the cumulative frequency of bleeding events increased proportionally to the increase in the follow-up time. Bruising events were the most frequently reported bleeding events (17.4%). Intracranial and gastrointestinal bleeding events were reported in 0.3% and 3.5% of patients respectively.
In the comparative period of the phase 3 acute GvHD study, bleeding events were reported in 25.0%and 22.0% of patients in the ruxolitinib and BAT arms respectively. The sub-groups of bleeding events were generally similar between treatment arms: bruising events (5.9% in ruxolitinib vs. 6.7% in BAT arm), gastrointestinal events (9.2% vs. 6.7%) and other haemorrhage events (13.2% vs. 10.7%). Intracranial bleeding events were reported in 0.7% of patients in the BAT arm and in no patients in the ruxolitinib arm.
In the comparative period of the phase 3 chronic GvHD study, bleeding events were reported in 11.5% and 14.6% of patients in the ruxolitinib and BAT arms respectively. The sub-groups of bleeding events were generally similar between treatment arms: bruising events (4.2% in ruxolitinib vs. 2.5% in BAT arm), gastrointestinal events (1.2% vs. 3.2%) and other haemorrhage events (6.7% vs. 10.1%).No intracranial bleeding events were reported in either treatment arm.
Infections: In the phase 3 pivotal studies in MF, grade 3 or 4 urinary tract infection was reported in 1.0% of patients, herpes zoster in 4.3% and tuberculosis in 1.0%. In phase 3 clinical studies sepsis was reported in 3.0% of patients. An extended follow-up of patients treated with ruxolitinib showed no trends towards an increase in the rate of sepsis over time.
In the randomised period of the phase 3 studies in PV patients, one (0.5%) CTCAE grade 3 and no grade 4 urinary tract infection was reported. The rate of herpes zoster was similar in PV (4.3%) patients and MF (4.0%) patients. There was one report of CTCAE grade 3 post-herpetic neuralgia amongst the PV patients. Pneumonia was reported in 0.5% of patients treated with ruxolitinib compared to 1.6% of patients in reference treatments. No patients in the ruxolitinib arm reported sepsis or tuberculosis.
During long-term follow-up of phase 3 studies in PV, frequently reported infections were urinary tract infections (11.8%), herpes zoster (14.7%) and pneumonia (7.1%). Sepsis was reported in 0.6% of patients. No patients reported tuberculosis in long-term follow-up.
In the phase 3 acute GvHD study, during the comparative period, urinary tract infections were reported in 9.9% (grade ≥3, 3.3%) of patients in the ruxolitinib arm compared to 10.7% (grade ≥3, 6.0%) in the BAT arm. CMV infections were reported in 28.3% (grade ≥3, 9.3%) of patients in the ruxolitinib arm compared to 24.0% (grade ≥3, 10.0%) in the BAT arm. Sepsis events were reported in 12.5% (grade ≥3, 11.1%) of patients in the ruxolitinib arm compared to 8.7% (grade ≥3, 6.0%) in the BAT arm. BK virus infection was reported only in the ruxolitinib arm in 3 patients with one grade 3 event. During extended follow-up of patients treated with ruxolitinib, urinary tract infections were reported in 17.9% (grade ≥3, 6.5%) of patients and CMV infections were reported in 32.3% (grade ≥3,11.4%) of patients. CMV infection with organ involvement was seen in very few patients; CMV colitis, CMV enteritis and CMV gastrointestinal infection of any grade were reported in four, two and one patients, respectively. Sepsis events, including septic shock, of any grade were reported in 25.4%(grade ≥3, 21.9%) of patients.
In the phase 3 chronic GvHD study, during the comparative period, urinary tract infections were reported in 8.5% (grade ≥3, 1.2%) of patients in the ruxolitinib arm compared to 6.3% (grade ≥3,1.3%) in the BAT arm. BK virus infection was reported in 5.5% (grade ≥3, 0.6%) of patients in the ruxolitinib arm compared to 1.3% in the BAT arm. CMV infections were reported in 9.1% (grade ≥3, 1.8%) of patients in the ruxolitinib arm compared to 10.8% (grade ≥3, 1.9%) in the BAT arm. Sepsis events were reported in 2.4% (grade ≥3, 2.4%) of patients in the ruxolitinib arm compared to 6.3% (grade ≥3, 5.7%) in the BAT arm. During extended follow-up of patients treated with ruxolitinib, urinary tract infections and BK virus infections were reported in 9.3% (grade ≥3, 1.3%) and 4.9% (grade ≥3, 0.4%) of patients, respectively. CMV infections and sepsis events were reported in 8.8% (grade ≥3, 1.3%) and 3.5% (grade ≥3, 3.5%) of patients, respectively.
Elevated lipase: In the randomised period of the RESPONSE study, the worsening of lipase values was higher in the ruxolitinib arm compared to the control arm, mainly due to the differences among grade 1 elevations (18.2% vs 8.1%). Grade ≥2 elevations were similar between treatment arms. In RESPONSE 2, the frequencies were comparable between the ruxolitinib and the control arm (10.8% vs 8%). During long-term follow-up of phase 3 PV studies, 7.4% and 0.9% of patients reported grade 3 and grade 4 elevation of lipase values. No concurrent signs and symptoms of pancreatitis with elevated lipase values were reported in these patients.
In phase 3 studies in MF, high lipase values were reported in 18.7% and 19.3% of patients in the ruxolitinib arms compared to 16.6% and 14.0% in the control arms in COMFORT-I and COMFORT-II studies, respectively. In patients with elevated lipase values, no concurrent signs and symptoms of pancreatitis were reported.
In the comparative period of the phase 3 acute GvHD study, new or worsened lipase values were reported in 19.7% of patients in the ruxolitinib arm compared to 12.5% in the BAT arm; corresponding grade 3 (3.1% vs 5.1%) and grade 4 (0% vs 0.8%) increases were similar. During extended follow-up of patients treated with ruxolitinib, increased lipase values were reported in 32.2% of patients; grade 3 and 4 were reported in 8.7% and 2.2% of patients respectively.
In the comparative period of the phase 3 chronic GvHD study, new or worsened lipase values were reported in 32.1% of patients in the ruxolitinib arm compared to 23.5% in the BAT arm; corresponding grade 3 (10.6% vs 6.2%) and grade 4 (0.6% vs 0%) increases were similar. During extended follow-up of patients treated with ruxolitinib, increased lipase values were reported in 35.9% of patients; grade 3 and 4 were observed in 9.5% and 0.4% of patients, respectively.
Increased systolic blood pressure: In the phase 3 pivotal clinical studies in MF an increase in systolic blood pressure of 20 mmHg or more from baseline was recorded in 31.5% of patients on at least one visit compared with 19.5% of the control-treated patients. In COMFORT-I (MF patients) the mean increase from baseline in systolic BP was 0-2 mmHg on ruxolitinib versus a decrease of 2-5 mmHg in the placebo arm. In COMFORT-II mean values showed little difference between the ruxolitinib-treated and the control-treated MF patients.
In the randomised period of the pivotal study in PV patients, the mean systolic blood pressure increased by 0.65 mmHg in the ruxolitinib arm versus a decrease of 2 mmHg in the BAT arm.
Paediatric patients: A total of 20 patients aged 12 to <18 years with GvHD were analysed for safety: 9 patients (5 in the ruxolitinib arm and 4 in the BAT arm) in the study REACH2 and 11 patients (4 in the ruxolitinib arm and 7 in the BAT arm) in the study REACH3. Based on the similar exposure observed in adolescents and adults, the safety of ruxolitinib at the recommended dose of 10 mg twice daily is similar in frequency and severity.
Elderly: A total of 29 patients in study REACH2 and 25 patients in REACH3 aged >65 years and treated with ruxolitinib were analysed for safety. Overall, no new safety concerns were identified and the safety profile in patients >65 years old is generally consistent with that of patients aged 18-65 years old.
View ADR Monitoring Form