Pharmacology: Mechanism of Action: Fingolimod is metabolized by sphingosine kinase to the active metabolite, fingolimod-phosphate. Fingolimod-phosphate is a sphingosine 1-phosphate receptor modulator, and binds with high affinity to sphingosine 1-phosphate receptors 1, 3, 4, and 5. Fingolimod-phosphate blocks the capacity of lymphocytes to egress from lymph nodes, reducing the number of lymphocytes in peripheral blood. The mechanism by which fingolimod exerts therapeutic effects in multiple sclerosis is unknown, but may involve reduction of lymphocyte migration into the central nervous system.
Pharmacodynamics: Heart Rate and Rhythm: Fingolimod causes a transient reduction in heart rate and AV conduction at treatment initiation (see PRECAUTIONS).
Heart rate progressively increases after the first day, returning to baseline values within 1 month of the start of chronic treatment.
Autonomic responses of the heart, including diurnal variation of heart rate and response to exercise, are not affected by fingolimod treatment.
Fingolimod treatment is not associated with a decrease in cardiac output.
Potential to Prolong the QT Interval: In a thorough QT interval study of doses of 1.25 or 2.5 mg fingolimod at steady-state, when a negative chronotropic effect of fingolimod was still present, fingolimod treatment resulted in a prolongation of QTc, with the upper boundary of the 90% confidence interval (CI) of 14.0 msec. There is no consistent signal of increased incidence of QTc outliers, either absolute or change from baseline, associated with fingolimod treatment. In MS studies, there was no clinically relevant prolongation of the QT interval, but patients at risk for QT prolongation were not included in clinical studies.
Immune System: Effects on Immune Cell Numbers in the Blood: In a study in which 12 adult subjects received Gilenya 0.5 mg daily, the lymphocyte count decreased to approximately 60% of baseline within 4 to 6 hours after the first dose. With continued daily dosing, the lymphocyte count continued to decrease over a 2-week period, reaching a nadir count of approximately 500 cells/mcL or approximately 30% of baseline. In a placebo-controlled study in 1272 MS patients (of whom 425 received fingolimod 0.5 mg daily and 418 received placebo), 18% (N=78) of patients on fingolimod 0.5 mg reached a nadir of <200 cells/mcL on at least 1 occasion. No patient on placebo reached a nadir of <200 cells/mcL. Low lymphocyte counts are maintained with chronic daily dosing of Gilenya 0.5 mg daily.
Chronic fingolimod dosing leads to a mild decrease in the neutrophil count to approximately 80% of baseline. Monocytes are unaffected by fingolimod.
Peripheral lymphocyte count increases are evident within days of stopping fingolimod treatment and typically normal counts are reached within 1 to 2 months.
Effect on Antibody Response: Gilenya reduces the immune response to vaccination, as evaluated in 2 studies.
In the first study, the immunogenicity of keyhole limpet hemocyanin (KLH) and pneumococcal polysaccharide vaccine (PPV-23) immunization were assessed by IgM and IgG titers in a steady-state, randomized, placebo-controlled study in healthy adult volunteers. Compared to placebo, antigen-specific IgM titers were decreased by 91% and 25% in response to KLH and PPV-23, respectively, in subjects on Gilenya 0.5 mg. Similarly, IgG titers were decreased by 45% and 50%, in response to KLH and PPV-23, respectively, in subjects on Gilenya 0.5 mg daily compared to placebo. The responder rate for Gilenya 0.5 mg as measured by the number of subjects with a >4-fold increase in KLH IgG was comparable to placebo and 25% lower for PPV-23 IgG, while the number of subjects with a >4 fold increase in KLH and PPV-23 IgM was 75% and 40% lower, respectively, compared to placebo. The capacity to mount a skin delayed-type hypersensitivity reaction to Candida and tetanus toxoid was decreased by approximately 30% in subjects on Gilenya 0.5 mg daily, compared to placebo. Immunologic responses were further decreased with fingolimod 1.25 mg (a dose higher than recommended in MS) (see PRECAUTIONS).
In the second study, the immunogenicity of Northern hemisphere seasonal influenza and tetanus toxoid vaccination was assessed in a 12-week steady-state, randomized, placebo-controlled study of Gilenya 0.5 mg in adult multiple sclerosis patients (n=136). The responder rate 3 weeks after vaccination, defined as seroconversion or a ≥4-fold increase in antibody directed against at least 1 of the 3 influenza strains, was 54% for Gilenya 0.5 mg and 85% in the placebo group. The responder rate 3 weeks after vaccination, defined as seroconversion or a ≥4-fold increase in antibody directed against tetanus toxoid was 40% for Gilenya 0.5 mg and 61% in the placebo group.
Pulmonary Function: Single fingolimod doses ≥5 mg (10-fold the recommended dose) are associated with a dose-dependent increase in airway resistance. In a 14-day study of 0.5, 1.25, or 5 mg/day, fingolimod was not associated with impaired oxygenation or oxygen desaturation with exercise or an increase in airway responsiveness to methacholine. Subjects on fingolimod treatment had a normal bronchodilator response to inhaled beta-agonists.
In a 14-day placebo-controlled study of adult patients with moderate asthma, no effect was seen for Gilenya 0.5 mg (recommended dose in MS). A 10% reduction in mean FEV1 at 6 hours after dosing was observed in adult patients receiving fingolimod 1.25 mg (a dose higher than recommended for use in MS) on Day 10 of treatment. Fingolimod 1.25 mg was associated with a 5-fold increase in the use of rescue short-acting beta-agonists.
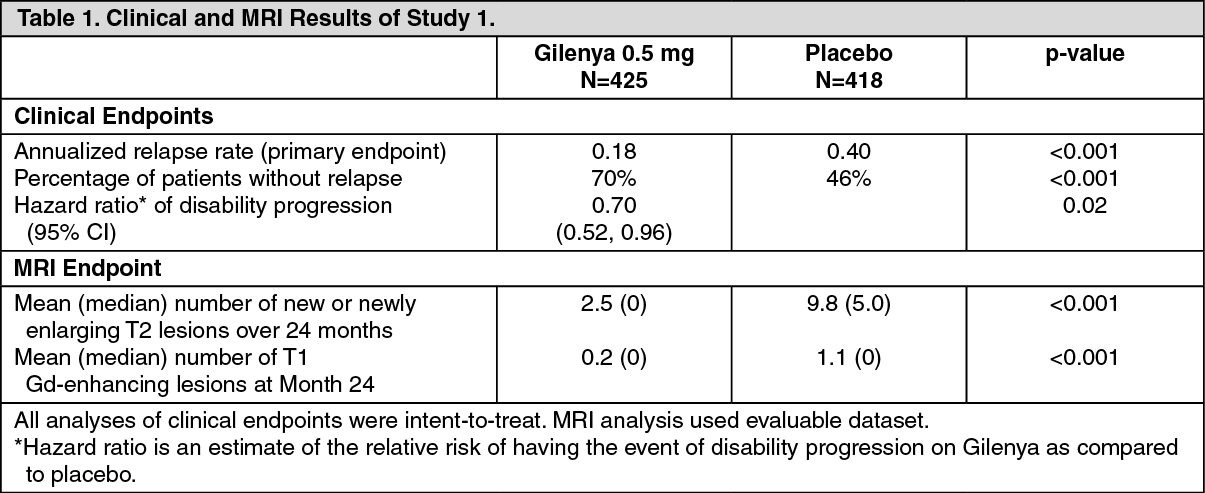
Clinical Studies: Adults: The efficacy of Gilenya was demonstrated in 2 studies that evaluated once-daily doses of Gilenya 0.5 mg and 1.25 mg in patients with relapsing-remitting MS (RRMS). Both studies included patients who had experienced at least 2 clinical relapses during the 2 years prior to randomization or at least 1 clinical relapse during the 1 year prior to randomization, and had an Expanded Disability Status Scale (EDSS) score from 0 to 5.5. Study 1 was a 2-year randomized, double-blind, placebo-controlled study in patients with RRMS who had not received any interferon-beta or glatiramer acetate for at least the previous 3 months and had not received any natalizumab for at least the previous 6 months. Neurological evaluations were performed at screening, every 3 months and at time of suspected relapse. MRI evaluations were performed at screening, Month 6, Month 12, and Month 24. The primary endpoint was the annualized relapse rate.
Median age was 37 years, median disease duration was 6.7 years and median EDSS score at baseline was 2.0. Patients were randomized to receive Gilenya 0.5 mg (N=425), 1.25 mg (N=429), or placebo (N=418) for up to 24 months. Median time on study drug was 717 days on 0.5 mg, 715 days on 1.25 mg and 719 days on placebo.
The annualized relapse rate was significantly lower in patients treated with Gilenya than in patients who received placebo. The secondary endpoint was the time to 3-month confirmed disability progression as measured by at least a 1-point increase from baseline in EDSS (0.5 point increase for patients with baseline EDSS of 5.5) sustained for 3 months. Time to onset of 3-month confirmed disability progression was significantly delayed with Gilenya treatment compared to placebo. The 1.25 mg dose resulted in no additional benefit over the Gilenya 0.5 mg dose. The results for this study are shown in Table 1 and figure. (See Table 1 and figure.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Study 2 was a 1-year randomized, double-blind, double-dummy, active-controlled study in patients with RRMS who had not received any natalizumab in the previous 6 months. Prior therapy with interferon-beta or glatiramer acetate up to the time of randomization was permitted.
Neurological evaluations were performed at screening, every 3 months, and at the time of suspected relapses. MRI evaluations were performed at screening and at Month 12. The primary endpoint was the annualized relapse rate.
Median age was 36 years, median disease duration was 5.9 years, and median EDSS score at baseline was 2.0. Patients were randomized to receive Gilenya 0.5 mg (N=431), 1.25 mg (N=426), or interferon beta-1a, 30 mcg via the intramuscular route (IM) once weekly (N=435) for up to 12 months. Median time on study drug was 365 days on Gilenya 0.5 mg, 354 days on 1.25 mg, and 361 days on interferon beta-1a IM.
The annualized relapse rate was significantly lower in patients treated with Gilenya 0.5 mg than in patients who received interferon beta-1a IM. The key secondary endpoints were number of new and newly enlarging T2 lesions and time to onset of 3-month confirmed disability progression as measured by at least a 1-point increase from baseline in EDSS (0.5 point increase for those with baseline EDSS of 5.5) sustained for 3 months. The number of new and newly enlarging T2 lesions was significantly lower in patients treated with Gilenya than in patients who received interferon beta-1a IM. There was no significant difference in the time to 3-month confirmed disability progression between Gilenya and interferon beta 1a-treated patients at 1 year. The 1.25 mg dose resulted in no additional benefit over the Gilenya 0.5 mg dose. The results for this study are shown in Table 2. (See Table 2.)
Click on icon to see table/diagram/image
Pooled results of study 1 and study 2 showed a consistent and statistically significant reduction of annualized relapse rate compared to comparator in subgroups defined by gender, age, prior MS therapy, and disease activity.
Pediatric Patients (10 to less than 18 Years of Age): Study 4 (NCT 01892722) evaluated the efficacy of once-daily oral doses of GILENYA 0.25 mg or GILENYA 0.5 mg in pediatric patients 10 to less than 18 years of age with relapsing-remitting multiple sclerosis. Study 4 was a 215-patient, double-blind, randomized, clinical trial that compared GILENYA to intramuscular interferon beta-1a. Prior therapy with interferon-beta, dimethyl fumarate, or glatiramer acetate up to the time of randomization was permitted. The study included patients who had experienced at least 1 clinical relapse during the year prior or 2 relapses during the 2 years prior to screening, or evidence of 1 or more Gd enhancing lesions on MRI within 6 months prior to randomization, and had an EDSS score from 0 to 5.5. Neurological evaluations were scheduled at screening, every 3 months, and at the time of suspected relapses. MRI evaluations were performed at screening and every 6 months throughout the study. The primary endpoint was the annualized relapse rate.
At baseline, the median age was 16 years, median disease duration since first symptom was 1.5 years, and median EDSS score was 1.5. One patient received no study drug and is excluded from the analysis of efficacy. Median duration of exposure to study drug was 634 days in the GILENYA group (n = 107) and 547 days in the interferon beta-1a group (n = 107). In the GILENYA group, 6.5% of patients did not complete the study, compared to 18.5% in the interferon beta-1a group.
The primary endpoint, the annualized relapse rate (ARR), was significantly lower in patients treated with GILENYA (0.122) than in patients who received interferon beta-1a (0.675). Relative reduction in ARR was 81.9%. The annualized rate of the number of new or newly enlarged T2 lesions up to month 24 (key secondary endpoint) was significantly lower in patients treated with GILENYA, as was the number of Gd-enhancing T1 lesions per scan up to month 24.
Table 3 summarizes the results of Study 4. (See Table 3.)
Click on icon to see table/diagram/image
Pharmacokinetics: Absorption: The T
max of fingolimod is 12-16 hours. The apparent absolute oral bioavailability is 93%.
Food intake does not alter C
max or (AUC) of fingolimod or fingolimod-phosphate. Therefore Gilenya may be taken without regard to meals.
Steady-state blood concentrations are reached within 1 to 2 months following once-daily administration and steady-state levels are approximately 10-fold greater than with the initial dose.
Distribution: Fingolimod highly (86%) distributes in red blood cells. Fingolimod-phosphate has a smaller uptake in blood cells of <17%. Fingolimod and fingolimod-phosphate are >99.7% protein bound. Fingolimod and fingolimod-phosphate protein binding is not altered by renal or hepatic impairment.
Fingolimod is extensively distributed to body tissues with a volume of distribution of about 1200±260 L.
Metabolism: The biotransformation of fingolimod in humans occurs by 3 main pathways: by reversible stereoselective phosphorylation to the pharmacologically active
(S)-enantiomer of fingolimod-phosphate, by oxidative biotransformation catalyzed mainly by the cytochrome P450 4F2 (CYP4F2) and possibly other CYP4F isoenzymes with subsequent fatty acid-like degradation to inactive metabolites, and by formation of pharmacologically inactive non-polar ceramide analogs of fingolimod.
Inhibitors or inducers of CYP4F2 and possibly other CYP4F isozymes might alter the exposure of fingolimod or fingolimod-phosphate. In vitro studies in hepatocytes indicated that CYP3A4 may contribute to fingolimod metabolism in the case of strong induction of CYP3A4.
Following single oral administration of [
14C] fingolimod, the major fingolimod-related components in blood, as judged from their contribution to the AUC up to 816 hours post-dose of total radiolabeled components, are fingolimod itself (23.3%), fingolimod-phosphate (10.3%), and inactive metabolites [M3 carboxylic acid metabolite (8.3%), M29 ceramide metabolite (8.9%), and M30 ceramide metabolite (7.3%)].
Elimination: Fingolimod blood clearance is 6.3±2.3 L/h, and the average apparent terminal half-life (t
½) is 6 to 9 days. Blood levels of fingolimod-phosphate decline in parallel with those of fingolimod in the terminal phase, yielding similar half-lives for both.
After oral administration, about 81% of the dose is slowly excreted in the urine as inactive metabolites. Fingolimod and fingolimod-phosphate are not excreted intact in urine but are the major components in the feces with amounts of each representing less than 2.5% of the dose.
Specific Populations: Pediatric Patients: The median fingolimod-phosphate (fingolimod-P) concentration in pediatric MS patients aged 10 to less than 18 years was 1.10 ng/mL, as compared to 1.35 ng/mL in adult MS patients.
Geriatric Patients: The mechanism for elimination and results from population pharmacokinetics suggest that dose adjustment would not be necessary in elderly patients. However, clinical experience in patients aged above 65 years is limited.
Gender: Gender has no clinically significant influence on fingolimod and fingolimod-phosphate pharmacokinetics.
Race: The effects of race on fingolimod and fingolimod-phosphate pharmacokinetics cannot be adequately assessed due to a low number of non-white patients in the clinical program.
Renal Impairment: In adult patients with severe renal impairment, fingolimod C
max and AUC are increased by 32% and 43%, respectively, and fingolimod-phosphate C
max and AUC are increased by 25% and 14%, respectively, with no change in apparent elimination half-life. Based on these findings, the Gilenya 0.5 mg dose is appropriate for use in adult patients with renal impairment. Gilenya 0.25 mg and 0.5 mg are appropriate for use in pediatric patients with renal impairment. The systemic exposure of 2 metabolites (M2 and M3) is increased by 3- and 13-fold, respectively. The toxicity of these metabolites has not been fully characterized.
A study in patients with mild or moderate renal impairment has not been conducted.
Hepatic Impairment: In subjects with mild, moderate, or severe hepatic impairment (Child-Pugh class A, B, and C), no change in fingolimod C
max was observed, but fingolimod AUC
0-∞ was increased respectively by 12%, 44%, and 103%. In patients with severe hepatic impairment (Child-Pugh class C), fingolimod-phosphate C
max was decreased by 22% and AUC
0-96 hours was decreased by 29%. The pharmacokinetics of fingolimod-phosphate was not evaluated in patients with mild or moderate hepatic impairment. The apparent elimination half-life of fingolimod is unchanged in subjects with mild hepatic impairment, but is prolonged by about 50% in patients with moderate or severe hepatic impairment.
Patients with severe hepatic impairment (Child-Pugh class C) should be closely monitored, as the risk of adverse reactions is greater (see PRECAUTIONS).
No dose adjustment is needed in patients with mild or moderate hepatic impairment (Child-Pugh class A and B).
Drug Interactions: Ketoconazole: The coadministration of ketoconazole (a potent inhibitor of CYP3A and CYP4F) 200 mg twice daily at steady-state and a single dose of fingolimod 5 mg led to a 70% increase in AUC of fingolimod and fingolimod-phosphate. Patients who use Gilenya and systemic ketoconazole concomitantly should be closely monitored, as the risk of adverse reactions is greater (see INTERACTIONS).
Carbamazepine: The coadministration of carbamazepine (a potent CYP450 enzyme inducer) 600 mg twice daily at steady-state and a single dose of fingolimod 2 mg decreased blood concentrations (AUC) of fingolimod and fingolimod-phosphate by approximately 40%. The clinical impact of this decrease is unknown.
Other strong CYP450 enzyme inducers, e.g., rifampicin, phenytoin, phenobarbital, and St. John's wort, may also reduce AUC of fingolimod and fingolimod-phosphate. The clinical impact of this potential decrease is unknown.
Potential of Fingolimod and Fingolimod-phosphate to Inhibit the Metabolism of Comedications: In vitro inhibition studies using pooled human liver microsomes and specific metabolic probe substrates demonstrate that fingolimod has little or no capacity to inhibit the activity of the following CYP enzymes: CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5, or CYP4A9/11 (fingolimod only), and similarly fingolimod-phosphate has little or no capacity to inhibit the activity of CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, or CYP3A4 at concentrations up to 3 orders of magnitude of therapeutic concentrations. Therefore, fingolimod and fingolimod-phosphate are unlikely to reduce the clearance of drugs that are mainly cleared through metabolism by the major CYP isoenzymes described previously.
Potential of Fingolimod and Fingolimod-phosphate to Induce its Own and/or the Metabolism of Comedications: Fingolimod was examined for its potential to induce human CYP3A4, CYP1A2, CYP4F2, and MDR1 (P-glycoprotein) mRNA and CYP3A, CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, and CYP4F2 activity in primary human hepatocytes. Fingolimod did not induce mRNA or activity of the different CYP enzymes and MDR1 with respect to the vehicle control; therefore, no clinically relevant induction of the tested CYP enzymes or MDR1 by fingolimod are expected at therapeutic concentrations. Fingolimod-phosphate was also examined for its potential to induce mRNA and/or activity of human CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP3A, CYP4F2, CYP4F3B, and CYP4F12. Fingolimod-phosphate is not expected to have clinically significant induction effects on these enzymes at therapeutic doses of fingolimod. In vitro experiments did not provide an indication of CYP induction by fingolimod-phosphate.
Transporters: Based on in vitro data, fingolimod as well as fingolimod-phosphate are not expected to inhibit the uptake of comedications and/or biologics transported by the organic anion transporting polypeptides OATP1B1, OATP1B3, or the sodium taurocholate co-transporting polypeptide (NTCP). Similarly, they are not expected to inhibit the efflux of comedications and/or biologics transported by the breast cancer resistance protein (BCRP), the bile salt export pump (BSEP), the multidrug resistance-associated protein 2 (MRP2), or P-glycoprotein (P-gp) at therapeutic concentrations.
Oral Contraceptives: The coadministration of fingolimod 0.5 mg daily with oral contraceptives (ethinylestradiol and levonorgestrel) did not elicit any clinically significant change in oral contraceptives exposure. Fingolimod and fingolimod-phosphate exposure were consistent with those from previous studies. No interaction studies have been performed with oral contraceptives containing other progestagens; however, an effect of fingolimod on their exposure is not expected.
Cyclosporine: The pharmacokinetics of single-dose fingolimod was not altered during coadministration with cyclosporine at steady-state, nor was cyclosporine steady-state pharmacokinetics altered by fingolimod. These data indicate that Gilenya is unlikely to reduce or increase the clearance of drugs cleared mainly by CYP3A4. Potent inhibition of transporters MDR1 (P-gp), MRP2, and OATP-1B1 does not influence fingolimod disposition.
Isoproterenol, Atropine, Atenolol, and Diltiazem: Single-dose fingolimod and fingolimod-phosphate exposure was not altered by coadministered isoproterenol or atropine. Likewise, the single-dose pharmacokinetics of fingolimod and fingolimod-phosphate and the steady-state pharmacokinetics of both atenolol and diltiazem were unchanged during the coadministration of the latter 2 drugs individually with fingolimod.
Population Pharmacokinetics Analysis: A population pharmacokinetics evaluation performed in MS patients did not provide evidence for a significant effect of fluoxetine and paroxetine (strong CYP2D6 inhibitors) on fingolimod or fingolimod-phosphate predose concentrations. In addition, the following commonly coprescribed substances had no clinically relevant effect (<20%) on fingolimod or fingolimod-phosphate predose concentrations: baclofen, gabapentin, oxybutynin, amantadine, modafinil, amitriptyline, pregabalin, and corticosteroids.
Nonclinical Toxicology: Carcinogenesis, Mutagenesis, Impairment of Fertility: Oral carcinogenicity studies of fingolimod were conducted in mice and rats. In mice, fingolimod was administered at oral doses of 0, 0.025, 0.25, and 2.5 mg/kg/day for up to 2 years. The incidence of malignant lymphoma was increased in males and females at the mid and high dose. The lowest dose tested (0.025 mg/kg/day) is less than the RHD of 0.5 mg/day on a body surface area (mg/m
2) basis. In rats, fingolimod was administered at oral doses of 0, 0.05, 0.15, 0.5, and 2.5 mg/kg/day. No increase in tumors was observed. The highest dose tested (2.5 mg/kg/day) is approximately 50 times the RHD on a mg/m
2 basis.
Fingolimod was negative in a battery of
in vitro (Ames, mouse lymphoma thymidine kinase, chromosomal aberration in mammalian cells) and
in vivo (micronucleus in mouse and rat) assays.
When fingolimod was administered orally (0, 1, 3, and 10 mg/kg/day) to male and female rats prior to and during mating, and continuing to Day 7 of gestation in females, no effect on fertility was observed up to the highest dose tested (10 mg/kg), which is approximately 200 times the RHD on a mg/m
2 basis.
Animal Toxicology and/or Pharmacology: Lung toxicity was observed in 2 different strains of rats and in dogs and monkeys. The primary findings included increase in lung weight, associated with smooth muscle hypertrophy, hyperdistention of the alveoli, and/or increased collagen. Insufficient or lack of pulmonary collapse at necropsy, generally correlated with microscopic changes, was observed in all species. In rats and monkeys, lung toxicity was observed at all oral doses tested in chronic studies. The lowest doses tested in rats (0.05 mg/kg/day in the 2-year carcinogenicity study) and monkeys (0.5 mg/kg/day in the 39-week toxicity study) are similar to and approximately 20 times the RHD on a mg/m
2 basis, respectively.
In the 52-week oral study in monkeys, respiratory distress associated with ketamine administration was observed at doses of 3 and 10 mg/kg/day; the most affected animal became hypoxic and required oxygenation. As ketamine is not generally associated with respiratory depression, this effect was attributed to fingolimod. In a subsequent study in rats, ketamine was shown to potentiate the bronchoconstrictive effects of fingolimod. The relevance of these findings to humans is unknown.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image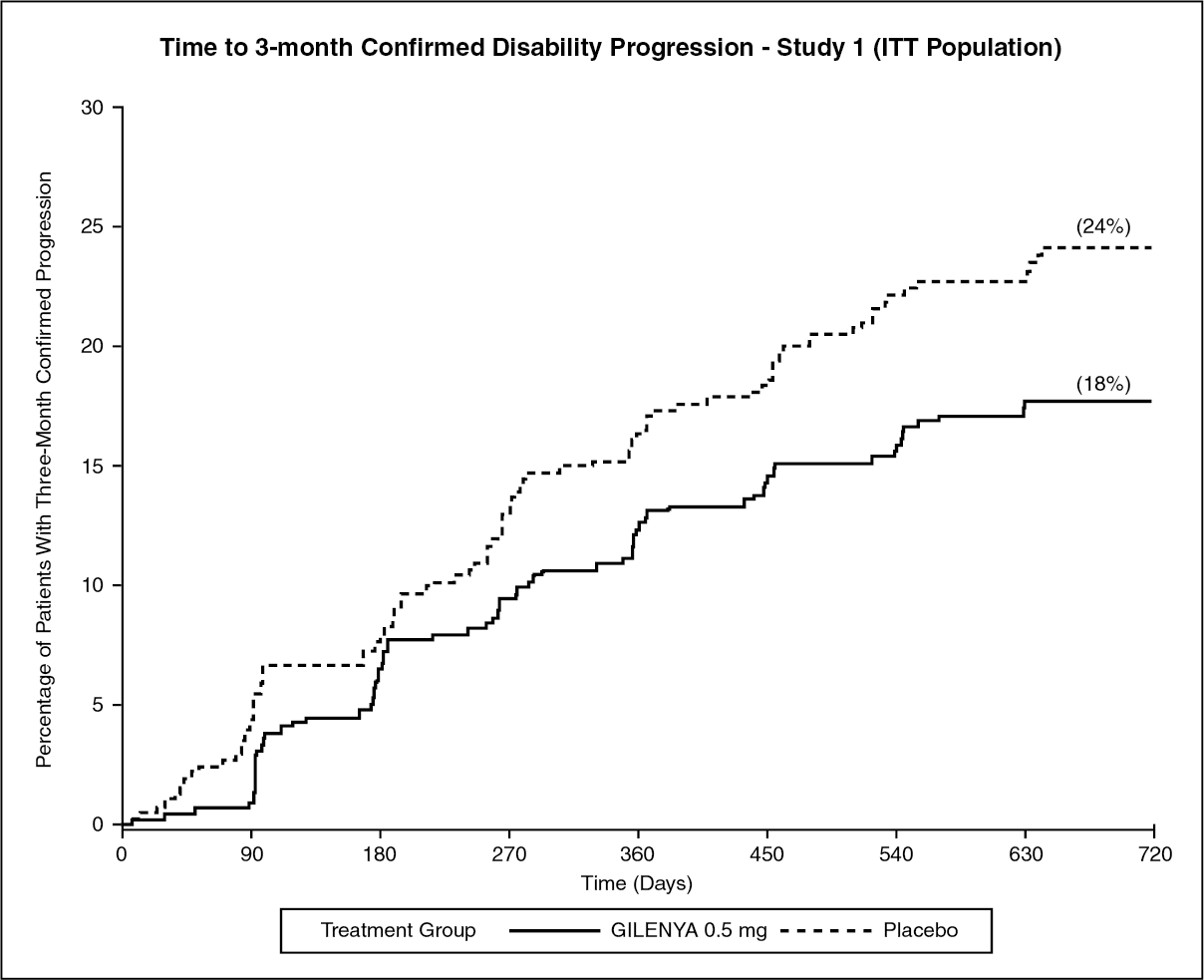 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image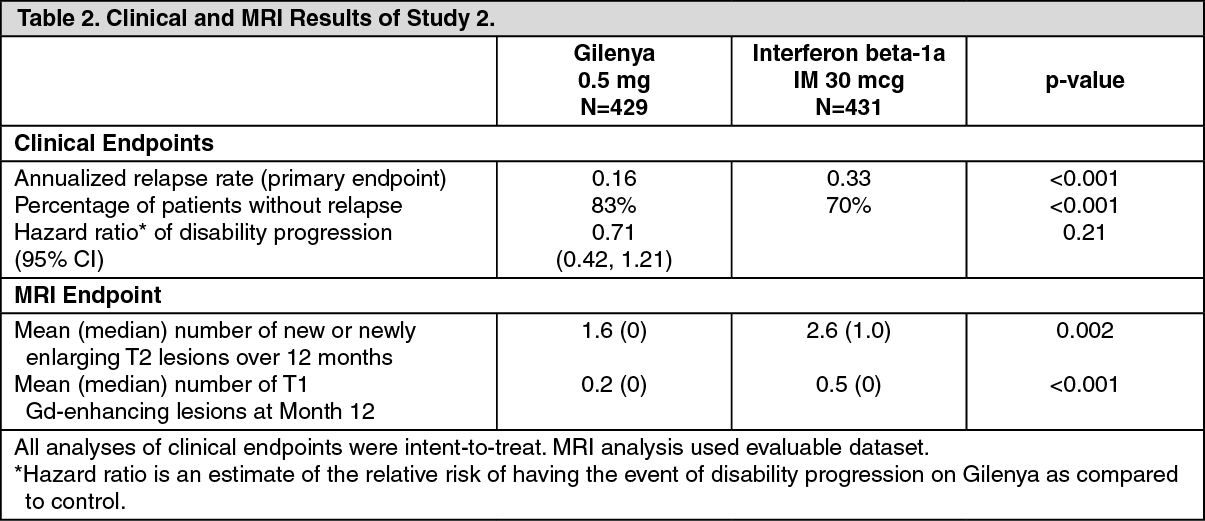 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image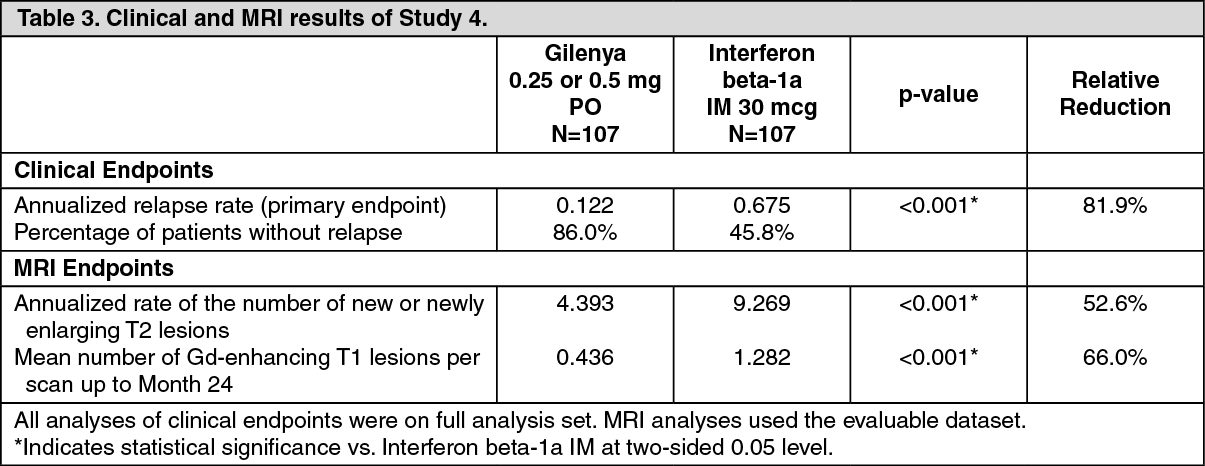 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image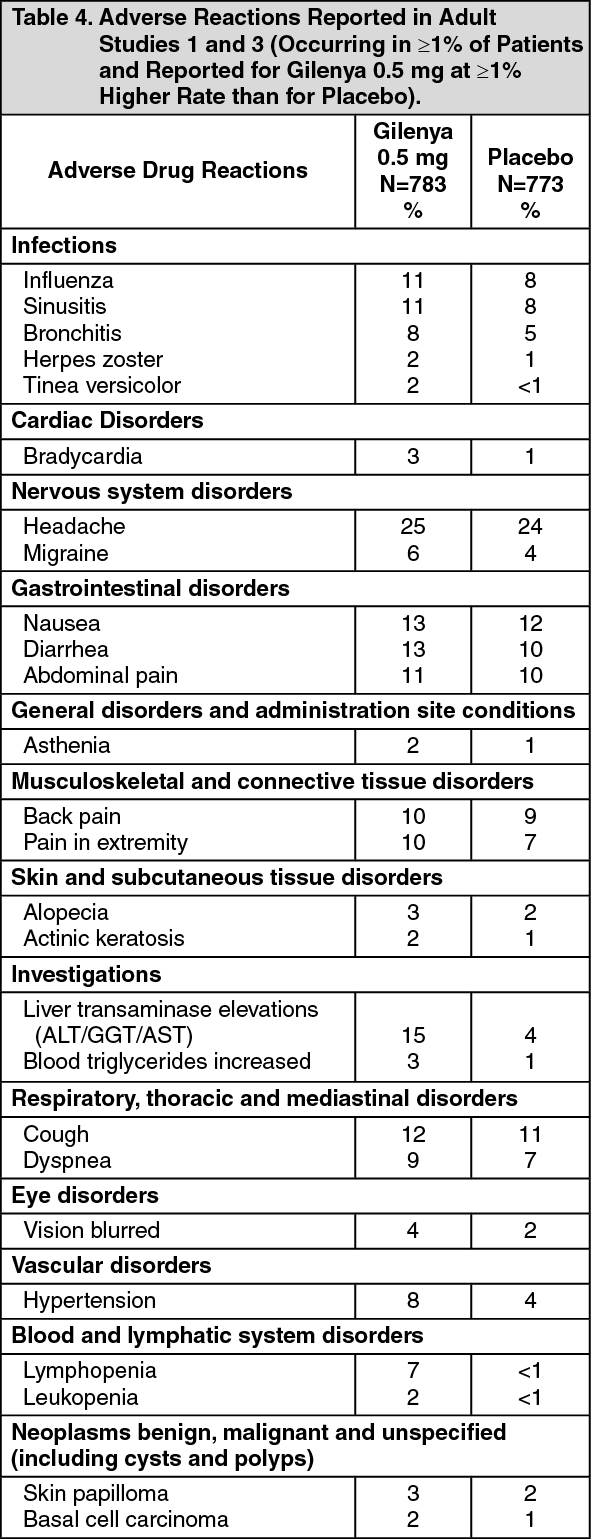 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
