Pharmacotherapeutic group: Urinary antispasmodics.
ATC code: G04B D07.
Pharmacology: Pharmacodynamics: Mechanism of action: Tolterodine is a competitive, specific muscarinic receptor antagonist with a selectivity for the urinary bladder over salivary glands
in vivo.
Pharmacodynamic effects: One of the tolterodine metabolites (5-hydroxymethyl derivative) exhibits a pharmacological profile similar to that of the parent compound. In extensive metabolisers this metabolite contributes significantly to the therapeutic effect (see Pharmacokinetics as follows).
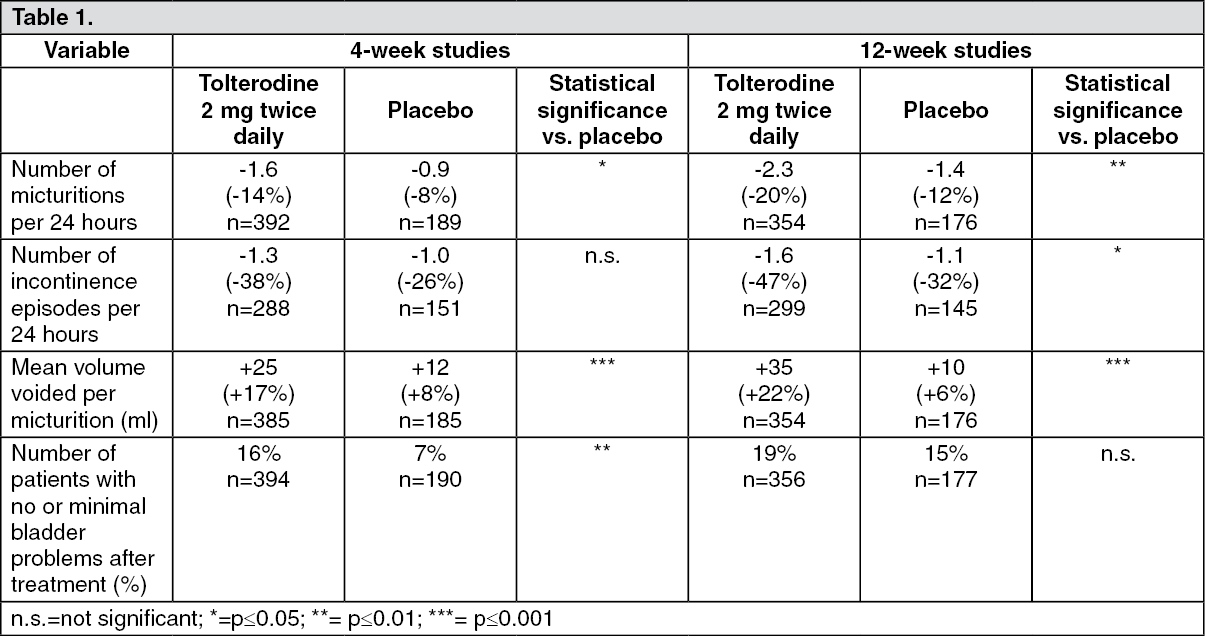
Clinical efficacy and safety: Effect of the treatment can be expected within 4 weeks.
Effect of treatment with tolterodine 2 mg twice daily after 4 and 12 weeks, respectively, compared with placebo (pooled data). Absolute change and percentage change relative to baseline. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The effect of tolterodine was evaluated in patients, examined with urodynamic assessment at baseline and, depending on the urodynamic result, they were allocated to a urodynamic positive (motor urgency) or a urodynamic negative (sensory urgency) group. Within each group, the patients were randomised to receive either tolterodine or placebo. The study could not provide convincing evidence that tolterodine had effects over placebo in patients with sensory urgency.
The clinical effects of tolterodine on QT interval were studied in ECGs obtained from over 600 treated patients, including the elderly and patients with pre-existing cardiovascular disease. The changes in QT intervals did not significantly differ between placebo and treatment groups.
The effect of tolterodine on QT-prolongation was investigated further in 48 healthy male and female volunteers aged 18-55 years. Subjects were administered 2 mg twice daily and 4 mg twice daily tolterodine as the immediate release formulations. The results (Fridericia corrected) at peak tolterodine concentration (1 hour) showed mean QTc interval increases of 5.0 and 11.8 msec for tolterodine doses of 2 mg twice daily and 4 mg twice daily respectively and 19.3 msec for moxifloxacin (400 mg) which was used as an active, internal control. A pharmacokinetic/pharmacodynamic model estimated that QTc interval increases in poor metabolisers (devoid of CYP2D6) treated with tolterodine 2 mg twice daily are comparable to those observed in extensive metabolisers receiving 4 mg twice daily. At both doses of tolterodine, no subject, irrespective of their metabolic profile, exceeded 500 msec for absolute QTcF or 60 msec for change from baseline that are considered thresholds of particular concern. The 4 mg twice daily dose corresponds to a peak exposure (C
max) of three times that obtained with the highest therapeutic dose of tolterodine extended release capsules.
Pharmacokinetics: Pharmacokinetic characteristics specific for this formulation: Tolterodine is rapidly absorbed. Both tolterodine and the 5-hydroxymethyl metabolite reach maximal serum concentrations 1-3 hours after dose. The half-life for tolterodine given as the tablet is 2-3 hours in extensive and about 10 hours in poor metabolisers (devoid of CYP2D6). Steady state concentrations are reached within 2 days after administration of the tablets.
Food does not influence the exposure to the unbound tolterodine and the active 5-hydroxymethyl metabolite in extensive metabolisers, although the tolterodine levels increase when taken with food. Clinically relevant changes are likewise not expected in poor metabolisers.
Absorption: After oral administration, tolterodine is subject to CYP2D6 catalysed first-pass metabolism in the liver, resulting in the formation of the 5-hydroxymethyl derivative, a major pharmacologically equipotent metabolite.
The absolute bioavailability of tolterodine is 17% in extensive metabolisers, the majority of the patients, and 65% in poor metabolisers (devoid of CYP2D6).
Distribution: Tolterodine and the 5-hydroxymethyl metabolite bind primarily to orosomucoid. The unbound fractions are 3.7% and 36%, respectively. The volume of distribution of tolterodine is 113 L.
Elimination: Tolterodine is extensively metabolised by the liver following oral dosing. The primary metabolic route is mediated by the polymorphic enzyme CYP2D6 and leads to the formation of the 5-hydroxymethyl metabolite. Further metabolism leads to formation of the 5-carboxylic acid and N-dealkylated 5-carboxylic acid metabolites, which account for 51% and 29% of the metabolites recovered in the urine, respectively. A subset (about 7%) of the population is devoid of CYP2D6 activity. The identified pathway of metabolism for these individuals (poor metabolisers) is dealkylation via CYP3A4 to N-dealkylated tolterodine, which does not contribute to the clinical effect. The remainder of the population is referred to as extensive metabolisers. The systemic clearance of tolterodine in extensive metabolisers is about 30 L/h. In poor metabolisers, the reduced clearance leads to significantly higher serum concentrations of tolterodine (about 7-fold) and negligible concentrations of the 5-hydroxymethyl metabolite are observed.
The 5-hydroxymethyl metabolite is pharmacologically active and equipotent with tolterodine. Because of the differences in the protein-binding characteristics of tolterodine and the 5-hydroxymethyl metabolite, the exposure (AUC) of unbound tolterodine in poor metabolisers is similar to the combined exposure of unbound tolterodine and the 5-hydroxymethyl metabolite in patients with CYP2D6 activity given the same dosage regimen. The safety, tolerability and clinical response are similar irrespective of phenotype.
The excretion of radioactivity after administration of [
14C]-tolterodine is about 77% in urine and 17% in faeces. Less than 1% of the dose is recovered as unchanged drug, and about 4% as the 5-hydroxymethyl metabolite. The carboxylated metabolite and the corresponding dealkylated metabolite account for about 51% and 29% of the urinary recovery, respectively.
Linearity/non-linearity: The pharmacokinetics is linear in the therapeutic dosage range.
Hepatic impairment: About 2-fold higher exposure of unbound tolterodine and the 5-hydroxymethyl metabolite is found in subjects with liver cirrhosis (see Dosage & Administration and Precautions).
Impaired renal function: The mean exposure of unbound tolterodine and its 5-hydroxymethyl metabolite is doubled in patients with severe renal impairment (inulin clearance GFR ≤30 ml/min). The plasma levels of other metabolites were markedly (up to 12-fold) increased in these patients. The clinical relevance of the increased exposure of these metabolites is unknown. There is no data in mild to moderate renal impairment (see Dosage & Administration and Precautions).
Toxicology: Preclinical Safety Data: In toxicity, genotoxicity, carcinogenicity and safety pharmacology studies no clinically relevant effects have been observed, except those related to the pharmacological effect of the drug.
Reproduction studies have been performed in mice and rabbits.
In mice, there was no effect of tolterodine on fertility or reproductive function. Tolterodine produced embryo death and malformations at plasma exposures (C
max or AUC) 20 or 7 times higher than those seen in treated humans.
In rabbits, no malformative effect was seen, but the studies were conducted at 20 or 3 times higher plasma exposure (C
max or AUC) than those expected in treated humans.
Tolterodine, as well as its active human metabolites, prolong action potential duration (90% repolarisation) in canine purkinje fibres (14 - 75 times therapeutic levels) and block the K+-current in cloned human ether-a-go-go-related gene (hERG) channels (0,5 - 26,1 times therapeutic levels). In dogs, prolongation of the QT interval has been observed after application of tolterodine and its human metabolites (3,1 - 61,0 times therapeutic levels). The clinical relevance of these findings is unknown.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
