Daunorubicin hydrochloride.
Active Ingredient: daunorubicin hydrochloride.
Daunorubicin is available as: Powder for solution for injection containing 20 mg of daunorubicin hydrochloride.
Excipients/Inactive Ingredients: Mannitol.
Pharmacology: Pharmacodynamics: Daunorubicin is an antineoplastic drug that exerts its cytotoxic/antiproliferative effects through interference with a number of biochemical and biological functions in target cells. Although the precise mechanism(s) of action has not been fully elucidated, the drug appears mainly to inhibit DNA and DNA-dependent RNA synthesis by forming a complex with DNA, via intercalation between base pairs and uncoiling of the helix. Daunorubicin may also interfere with polymerase and topoisomerase II activities, with regulation of gene expression and with oxidation/reduction reactions (generating highly reactive/highly toxic free radicals). A direct interaction between daunorubicin and the cellular membrane yielding alterations on the cell surface double layer has also been speculated. Daunorubicin is maximally cytotoxic during the S phase, but the drug is not cycle- or phase-specific. Antibacterial and immunosuppressive properties have also been ascribed to daunorubicin.
No controlled paediatric studies have been conducted.
Pharmacokinetics: Absorption: Daunorubicin is not absorbed by the gastrointestinal tract. Since the drug is extremely irritating to tissues, it has to be administered by the IV route: under these conditions the absorption is expected to be complete (i.e., if no extravasation occurs).
Distribution: Daunorubicin is widely distributed into body tissues, with highest levels in the spleen, kidneys, lungs and heart. The drug enters into cells and binds to cellular components, particularly nucleic acids. There is no evidence that daunorubicin can cross the blood-brain barrier, but the drug apparently crosses the placenta.
Metabolism: The drug undergoes a rapid and extensive metabolism in the liver and other tissues, mainly by cytoplasmic aldo-keto reductases. One hour after drug administration the predominant plasma species is the active metabolite daunorubicinol (13-OH daunorubicin). Further metabolism via reduction cleavage of the glycosidic bond produces aglycones, which have little or no antiproliferative activity and are demethylated and conjugated via sulfate and glucuronide by microsomal enzymes.
Excretion: Following rapid IV administration, total plasma concentrations of daunorubicin and its metabolites decline in a triphasic fashion, while plasma concentrations of unchanged daunorubicin decline in a biphasic fashion. The half-life averages 45 minutes in the initial phase and 18.5 hours in the terminal phase. The daunorubicinol half-life exceeds the 24 hours. Daunorubicin and its metabolites are excreted in urine and bile (approximately 40% of the administered dose). Urinary excretion of the drug and its metabolites is reported to be 14% to 23% of the given dose, with most urinary excretion occurring within 3 days. After the first 24 hours, the drug is excreted in the urine mainly as daunorubicinol.
Toxicology: Preclinical safety data: The LD50 of daunorubicin is 17.3-20 and 13-15 in mice and rats, respectively and about 5 mg/kg for dogs. The main target organs after a single dose are the hemolymphopoietic system and, especially in dogs, the gastrointestinal tract.
The toxic effects after repeated administrations have been investigated in rabbits, dogs and monkeys. The main target organs of daunorubicin in these animal species resulted the hemolymphopoietic system, gastrointestinal tract, kidney, liver and reproductive organs. Subacute and cardiotoxicity studies indicate that daunorubicin is cardiotoxic in all the laboratory animals tested.
Daunorubicin is genotoxic in most of the in vitro and in vivo tests performed, toxic to the reproductive organs, embryotoxic in rats and rabbits and teratogenic in rats. There is no information available on the administration of daunorubicin in animals during the peri- and post-natal periods and it is not known whether the compound is excreted in breast milk. Daunorubicin, like other anthracyclines and cytotoxic drugs, is carcinogenic in rats. Toxicity studies show that extravasation of the drug cause tissue necrosis.
Acute myeloblastic leukemia: Daunorubicin is indicated for the treatment of all stages of this disease, either as a single agent or in combination with other antiblastic drugs. It is also the treatment of choice for promyelocytic leukemia.
Acute lymphoblastic leukemia: Daunorubicin is very active in inducing remission in this disease. However, due to its side effects and the availability of other forms of therapy, Daunorubicin is only indicated in those cases that are resistant to other drugs. The combination chemotherapy regimen of Daunorubicin, prednisolone and vincristine in the acute phase of the disease has proved successful.
Other tumours: Positive responses have been observed with Daunorubicin in neuroblastoma and rhabdomyosarcoma.
The single dose can vary from 0.5 to 3 mg/kg. The doses of 0.5-1 mg/kg can be repeated at intervals of 1 or more days; the doses of 2 mg/kg should be given at intervals of 4 or more days; the doses of 2.5-3 mg/kg, although rarely used, should be given only at intervals of 7-14 days. The number of injections required varies widely from patient to patient and should be prescribed for each individual case, depending on individual response and tolerability, taking into account blood chemistry and bone marrow conditions and any association with other antiblastic agents. In both adults and children it is recommended not to exceed the total dose of 20 mg/kg (see Precautions).
Hepatic impairment: The dosage of Daunorubicin should be reduced in patients who present impairment of liver function so as to avoid an increase in global toxicity.
It is recommended a dosage reduction in patients with the following chemical parameters in the blood: Daunorubicin should not be administered to patients with severe hepatic impairment (Child-Pugh Grade C [total score 10-15] - see Contraindications).
For patients with mild and moderate hepatic impairment (Child-Pugh Grade A [total score 5-6] and B [total score 7-9]), dose reductions are recommended based on the following serum bilirubin values: Bilirubin 1.2 to 3 mg/dL: one-half of recommended starting dose.
Bilirubin >3 mg/dL: one-fourth of recommended starting dose.
Renal impairment: For patients with moderate renal impairment, glomerular filtration rate (GFR) between 10-20 mL/min or serum creatinine approximately between 3.4-7.9 mg/dL, the daunorubicin dose should be reduced by one-half (see Contraindications).
Method of Administration: Daunorubicin is not active if taken orally and should not be administered by intramuscular or intrathecal route. Administration is to be effected only by intravenous injection; it is advisable to effect the injection into the tubing of an intravenous infusion of normal saline solution already in situ, after ascertaining that the needle is perfectly in the vein. This technique reduces the danger of the drug leaking out and ensures that the vein is well washed through after administration of the drug.
Daunorubicin must not be mixed with heparin since these drugs show chemical incompatibility and, in certain proportions, form a precipitate.
Paediatric population: Daunorubicin dosage for children (over 2 years of age) is usually calculated based on the body surface area and adjusted to meet individual requirements of each patient, on the basis of clinical response and the patient's haematological status. Courses may be repeated after 3 to 6 weeks.
Current specialized protocols and guidelines should be consulted for appropriate treatment regimen.
For children over 2 years of age the maximum cumulative dose is 300 mg/m2.
For children under 2 years of age (or with body surface area below 0.5 m2), the maximum cumulative dose is 10 mg/kg.
Acute overdosage with daunorubicin will result in severe myelosuppression (mainly leukopenia and thrombocytopenia), gastrointestinal toxic effects (mainly mucositis) and acute cardiac complications. Very high single doses of daunorubicin may be expected to cause acute myocardial degeneration (within 24 hours) and severe myelosuppression (within 10-14 days). Treatment should aim to support the patient during this period. Delayed cardiac failures have been observed with anthracyclines up to 6 months after an overdose. Patients have to be observed carefully: if signs of cardiac failure arise, they should be treated along conventional lines.
Hypersensitivity to daunorubicin or any other component of the product, other anthracyclines, or anthracenediones.
Persistent myelosuppression.
Presence of severe infections.
Severe hepatic (Child-Pugh Grade C [total score 10-15]) or renal function impairment (GFR <10 mL/min or serum creatinine >7.9 mg/dL).
Myocardial insufficiency.
Recent myocardial infarction.
Severe arrhythmias.
Previous treatments with maximum cumulative doses of daunorubicin, other anthracyclines and/or anthracenediones (see Precautions).
General: Daunorubicin should be administered only under the supervision of physicians experienced in the use of cytotoxic therapy.
Patients should recover from acute toxicities of prior cytotoxic treatment (such as stomatitis, neutropenia, thrombocytopenia, and generalized infections) before beginning treatment with daunorubicin.
Hematologic Toxicity: Evaluation of response based on bone marrow status cellularity is necessary to guide daunorubicin treatment: myelosuppression will occur in all patients given therapeutic doses of the drug. Hematologic profiles should be assessed before and during each cycle of therapy with daunorubicin, including differential white blood cell (WBC) counts: marked cytopenia is expected and requires careful monitoring.
The nadir of leukocytes and platelets usually occurs between 10 and 14 days following drug administration, but cell counts generally return to the pre-treatment levels during the third week. Thrombocytopenia and anemia may also occur. Clinical consequences of severe myelosuppression include fever, infection, sepsis/septicemia, septic shock, hemorrhage, tissue hypoxia, or death. During a course of therapy special attention should be devoted to patients with severe neutropenia and fever (febrile neutropenia), a condition that can be possibly followed by septicemia and death.
Secondary Leukemia: Secondary leukemia, with or without a preleukemic phase, has been reported in patients treated with anthracyclines, including daunorubicin. Secondary leukemia is more common when such drugs are given in combination with DNA-damaging antineoplastic agents, in combination with radiotherapy, when patients have been heavily pre-treated with cytotoxic drugs, or when doses of the anthracyclines have been escalated. These leukemias can have a 1- to 3-year latency period.
Cardiac Function: Cardiotoxicity is a risk of anthracycline treatment that may be manifested by early (i.e., acute) or late (i.e., delayed) events.
Early (i.e., acute) Events: Early cardiotoxicity of daunorubicin consists mainly of sinus tachycardia and/or electrocardiogram (ECG) abnormalities such as non-specific ST-T wave changes. Tachyarrhythmias, including premature ventricular contractions, as well as heart block have also been reported. These effects do not usually predict subsequent development of delayed cardiotoxicity, are rarely of clinical importance, and are generally not a consideration for discontinuation of daunorubicin treatment.
Late (i.e., delayed) Events: Delayed cardiotoxicity usually develops late in the course of therapy with daunorubicin or within 2 to 3 months after treatment termination, but later events (several months to years after completion of treatment) have also been reported. Delayed cardiomyopathy is manifested by reduced left ventricular ejection fraction (LVEF) and/or signs and symptoms of congestive heart failure (CHF) such as dyspnea, pulmonary edema, dependent edema, cardiomegaly and hepatomegaly, oliguria, ascites, pleural effusion and gallop rhythm. Life-threatening CHF is the most severe form of anthracycline-induced cardiomyopathy and represents the cumulative dose-limiting toxicity of the drug.
Cardiac function should be assessed before patients undergo treatment with daunorubicin and must be monitored throughout therapy to minimize the risk of incurring severe cardiac impairment. The risk may be decreased through regular monitoring of LVEF during the course of treatment with prompt discontinuation of daunorubicin at the first sign of impaired function. The appropriate quantitative method for repeated assessment of cardiac function (evaluation of LVEF) includes multi-gated radionuclide angiography (MUGA) or echocardiography (ECHO). A baseline cardiac evaluation with an ECG and either a MUGA scan or an ECHO is recommended, especially in patients with risk factors for increased cardiotoxicity. Repeated MUGA or ECHO determinations of LVEF should be performed, particularly with higher, cumulative anthracycline doses. The technique used for assessment should be consistent throughout follow-up.
The risk of developing congestive heart failure (CHF) increases - in the absence of other cardiac risk factors - when the total cumulative dose of daunorubicin exceeds 500-600 mg/m2 in adults, 300 mg/m2 in children more than 2 years of age, or 10 mg/kg in children less than 2 years of age; these doses should only be exceeded with extreme caution.
Risk factors for cardiac toxicity include active or dormant cardiovascular disease, prior or concomitant radiotherapy to the mediastinal/pericardial area, previous therapy with other anthracyclines or anthracenediones, and concomitant use of drugs with the ability to suppress cardiac contractility or cardiotoxic drugs (e.g., trastuzumab). Anthracyclines including daunorubicin should not be administered in combination with other cardiotoxic agents unless the patient's cardiac function is closely monitored. Patients receiving anthracyclines after stopping treatment with other cardiotoxic agents, especially those with long half-lives such as trastuzumab, may also be at an increased risk of developing cardiotoxicity. Under these conditions, a total cumulative dose of 400 mg/m2 in adults should be exceeded only with extreme caution.
Cardiac function must be carefully monitored in patients receiving high cumulative doses and in those with risk factors. However, cardiotoxicity with daunorubicin may occur at lower cumulative doses whether or not cardiac risk factors are present.
In infants and children there appears to be a greater susceptibility to anthracycline-induced cardiac toxicity, and a long-term periodic evaluation of cardiac function has to be performed.
It is probable that the toxicity of daunorubicin and other anthracyclines or anthracenediones is additive.
Gastrointestinal: Daunorubicin may cause nausea and vomiting. Severe nausea and vomiting may produce dehydration. Nausea and vomiting may be prevented or alleviated by the administration of appropriate antiemetic therapy.
Mucositis (mainly stomatitis, less often esophagitis) may occur in patients undergoing daunorubicin therapy. Mucositis/stomatitis generally appear early after drug administration and, if severe, may progress over a few days to mucosal ulcerations. Most patients recover from this adverse event by the third week of therapy.
Tumor Lysis Syndrome: Daunorubicin may induce hyperuricemia as a consequence of the extensive purine catabolism that accompanies rapid drug-induced lysis of neoplastic cells (tumor-lysis syndrome). Blood uric acid levels, potassium, calcium phosphate, and creatinine should be evaluated after initial treatment. Hydration, urine alkalinization, and prophylaxis with allopurinol to prevent hyperuricemia may minimize potential complications of tumor-lysis syndrome.
Effects at Site of Injection: Phlebosclerosis may result from an injection into a small vessel or from repeated injections into the same vein. Following the recommended administration procedures may minimize the risk of phlebitis/thrombophlebitis at the injection site (see Dosage & Administration).
Extravasation: Extravasation of daunorubicin during intravenous injection may produce local pain, severe tissue lesions (vesication, severe cellulitis) and necrosis. Should signs or symptoms of extravasation occur during intravenous administration of daunorubicin, the drug infusion should be immediately stopped.
Alopecia: Complete alopecia involving beard growth and the scalp, axillary and pubic hair occurs almost always with full doses of daunorubicin. This side-effect may cause distress to patients but is usually reversible, with regrowth of hair, which usually occurs within two to three months from the termination of therapy.
Immunosuppressant Effects/Increased Susceptibility to Infections: Administration of live or live-attenuated vaccines in patients immunocompromised by chemotherapeutic agents including daunorubicin may result in serious or fatal infections. Vaccination with a live vaccine should be avoided in patients receiving daunorubicin. Killed or inactivated vaccines may be administered; however, the response to such vaccines may be diminished.
Effects on ability to drive and use machines: There have been no reports explicitly relating to effects of daunorubicin treatment on the ability to drive or use machines.
Hepatic Function: The major route of elimination of daunorubicin is the hepatobiliary system. Serum total bilirubin should be evaluated before and during treatment with daunorubicin. Patients with elevated bilirubin may experience slower clearance of drug with an increase in overall toxicity. Lower doses are recommended in these patients (see Dosage & Administration). Patients with severe hepatic impairment must not receive daunorubicin (see Contraindications).
Renal Function: Renal impairment can also enhance the toxicity of the recommended doses of daunorubicin, and renal function should be evaluated before starting treatment with daunorubicin (see Dosage & Administration and Contraindications).
Impairment of Fertility: Daunorubicin could induce chromosomal damage in human spermatozoa. Men undergoing treatment with daunorubicin should use effective contraceptive methods.
Pregnancy: Like most other anticancer drugs, daunorubicin has shown teratogenic, mutagenic and carcinogenic potential in animals. According to experimental data, the drug must be considered as a potential cause of fetal malformations when administered to a pregnant woman. There are no adequate and well-controlled studies in pregnant women, although a few women who received daunorubicin during the second and third trimesters of pregnancy have delivered apparently normal infants.
As a general rule, it is recommended that daunorubicin not be administered to patients who are pregnant. If the drug is used during pregnancy, or if the patient becomes pregnant while receiving the drug, the woman should be informed of the potential hazard to the fetus. Women of child-bearing potential who have to undergo daunorubicin therapy should be apprised of the potential hazard to the fetus and should be advised to avoid becoming pregnant during treatment. Daunorubicin should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Lactation: It is not known whether daunorubicin is excreted in human milk. As a general rule, it is recommended that daunorubicin not be administered to mothers who are breast-feeding.
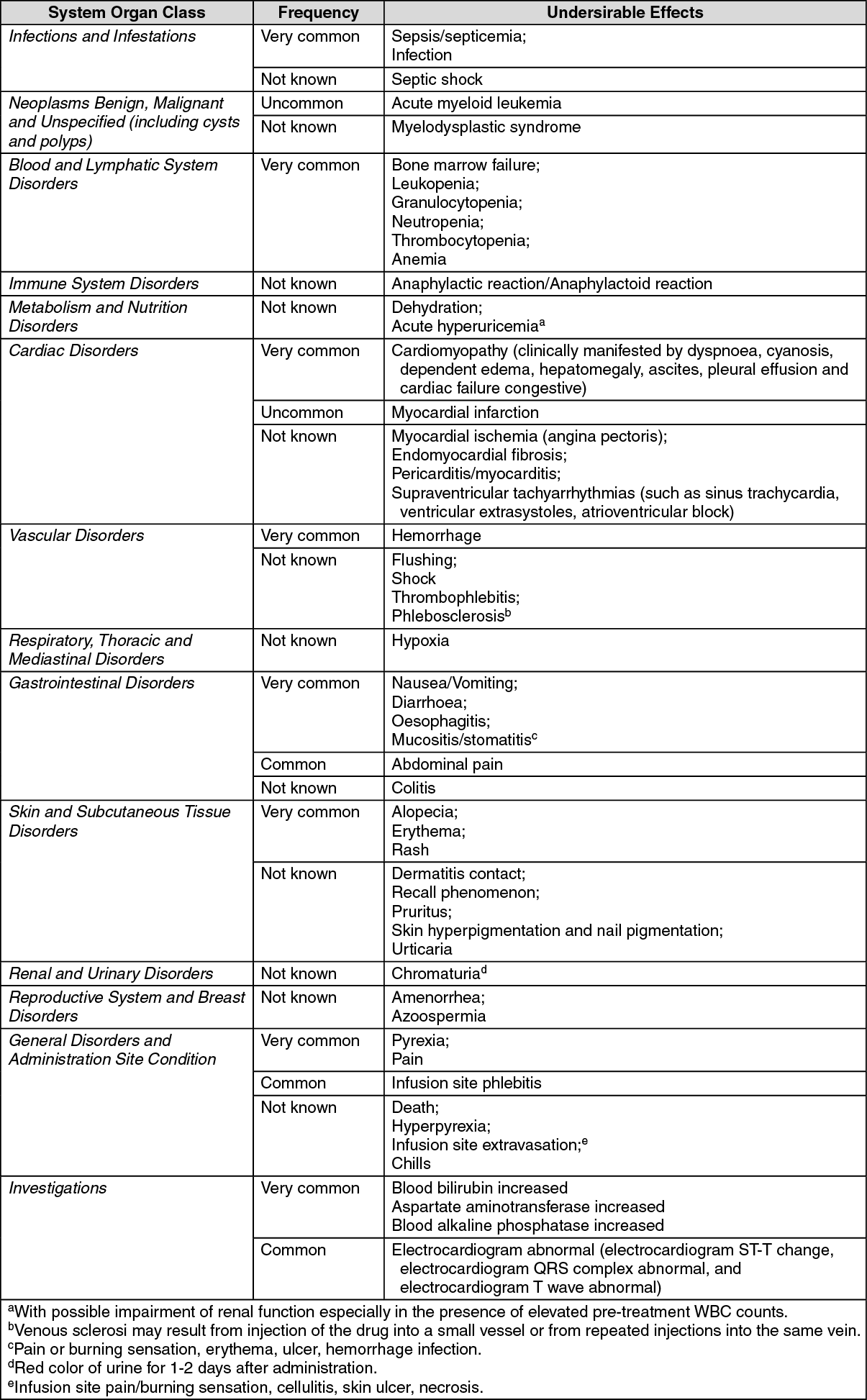
The adverse reactions are listed by system organ class, frequency category and grade of severity. Frequency categories are defined as: very common (≥1/10), common (≥1/100 to <1/10), uncommon (≥1/1,000 to <1/100), rare (≥1/10,000 to <1/1,000), very rare (<1/10,000), not known (cannot be estimated from the available data) (see also Precautions). (See table.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Daunorubicin is mainly used in combination with other cytotoxic drugs. Additive toxicity may occur especially with regard to bone marrow/haematologic and gastrointestinal effects (see Precautions). The use of daunorubicin in combination chemotherapy with other potentially cardiotoxic drugs, as well as the concomitant use of other cardioactive compounds (e.g., calcium channel blockers), requires monitoring of cardiac function throughout treatment. Changes in hepatic or renal function induced by concomitant therapies may affect daunorubicin metabolism, pharmacokinetics, therapeutic efficacy and/or toxicity.
Incompatibilities: Daunorubicin has been reported to be incompatible with heparin sodium, which might cause the drug to precipitate in the solution, and with aluminum. Incompatibility has also been reported when a daunorubicin hydrochloride solution is mixed with a solution of dexamethasone sodium phosphate, aztreonam, allopurinol sodium, fludarabine, piperacillin/tazobactam and aminophylline. Daunorubicin can be used in combination with other antitumor agents, but it is not recommended that it be mixed with other drugs in the same syringe.
Special precautions for disposal and other handling: Preparation of the solution: Daunorubicin hydrochloride powder for injection should be reconstituted by adding 4 mL of sterile Water for Injection. The vial content is under a negative pressure to minimize aerosol formation during reconstitution; particular care is needed when the needle is inserted: inhalation of any aerosol produced during reconstitution must be avoided. The vial should be gently shaken until the drug is completely dissolved. The resultant solution contains 5 mg of daunorubicin per mL. The reconstituted solution should be protected from light and is stable for 24 hours at room temperature or 48 hours at 4°C - 10°C.
Intravenous administration: The required dose of the reconstituted solution has to be withdrawn into a syringe containing 10 to 15 mL of 0.9% sodium chloride and slowly injected into the tubing of a freely flowing IV infusion of 0.9% sodium chloride or 5% dextrose to minimize the risk of drug extravasation and make sure that the vein is flushed after the administration of the drug.
Protective measures: The following protective recommendations are given due to the toxic nature of the compound: Personnel should be trained in good technique for reconstitution and handling;
Pregnant staff should be excluded from working with this drug;
Personnel handling daunorubicin should wear protective clothing: goggles, gowns and disposable gloves and masks;
A designated area should be defined for reconstitution (preferably under a laminar flow system). The work surface should be protected by disposable, plastic-backed, absorbent paper;
All items used for reconstitution, administration or cleaning, including gloves, should be placed in high-risk waste-disposal bags for high-temperature incineration;
Spillage or leakage should be treated with diluted sodium hypochlorite (1% available chlorine) solution, preferably by soaking, and then water;
All cleaning materials should be disposed of as indicated previously;
Accidental contact with the skin or eyes should be treated immediately by copious lavage with water, or soap and water, or sodium bicarbonate solution; medical attention should be sought;
Always wash hands after removing gloves;
The drug should be used within 24 hours of first penetration of the rubber stopper. Discard any unused solution.
Store below 30°C. Do not refrigerate/freeze.
Shelf life: 36 months.
L01DB02 - daunorubicin ; Belongs to the class of cytotoxic antibiotics, anthracyclines and related substances. Used in the treatment of cancer.
Daunoblastina powd for inj 20 mg
1's


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
