Therapeutic class: Irinotecan hydrochloride is an antineoplastic agent of the topoisomerase I inhibitor class, clinically investigated as CPT-11. Irinotecan is a semisynthetic derivative of camptothecin, an alkaloid extract from plants such as
Camptotheca acuminata, or is chemically synthesized.
Pharmacology: Pharmacodynamics: Mechanism of action: Irinotecan and its active metabolite SN-38 bind to the topoisomerase I - DNA complex and prevent re-ligation of these single-strand breaks. Current research suggests that the cytotoxicity of irinotecan is due to double-strand DNA damage produced during DNA synthesis when replication enzymes interact with the ternary complex formed by topoisomerase I, DNA, and either irinotecan or SN-38.
Irinotecan serves as a water-soluble precursor of the lipophilic metabolite SN-38. SN-38 is formed from irinotecan by carboxylesterase-mediated cleavage of the carbamate bond between the camptothecin moiety and the dipiperidino side chain. SN-38 is approximately 1000 times as potent as irinotecan as an inhibitor of topoisomerase I purified from human and rodent tumor cell lines.
In vitro cytotoxicity assays show that the potency of SN-38 relative to irinotecan varies from 2- to 2000-fold. However, the plasma area under the concentration versus time curve (AUC) values for SN-38 are 2% to 8% of irinotecan and SN-38 is 95% bound to plasma proteins compared to approximately 50% bound to plasma proteins for irinotecan. The precise contribution of SN-38 to the activity of irinotecan is thus unknown. Both irinotecan and SN-38 exist in an active lactone form and an inactive hydroxy acid anion form. A pH-dependent equilibrium exists between the two forms such that an acid pH promotes the formation of the lactone, while a more basic pH favors the hydroxy acid anion form.
Pharmacokinetics: Absorption and distribution: After intravenous infusion in humans, irinotecan plasma concentrations decline in a multiexponential manner, with a mean terminal elimination half-life of about 6 hours. The mean terminal elimination half-life of the active metabolite SN-38 is about 10 hours. The half-lives of the lactone (active) forms of irinotecan and SN-38 are similar to those of total irinotecan and SN-38, as the lactone and hydroxy acid forms are in equilibrium.
Over the dose range of 50 to 350 mg/m
2, the AUC of irinotecan increases linearly with dose; the AUC of SN-38 increases less than proportionally with dose. Maximum concentrations of the active metabolite SN-38 are generally seen within 1 hour following the end of a 90-minute infusion of irinotecan.
Irinotecan exhibits moderate plasma protein binding (30% to 68% bound). SN-38 is highly bound to human plasma proteins (approximately 95% bound). The plasma protein to which irinotecan and SN-38 predominantly binds is albumin.
Metabolism and excretion: Irinotecan (CPT-11) is subject to extensive metabolic conversion by various enzyme systems, including esterases to form the active metabolite SN-38, and UGT1A1 mediating glucuronidation of SN-38 to form the inactive glucuronide metabolite SN-38G. Irinotecan (CPT-11) can also undergo CYP3A4-mediated oxidative metabolism to several pharmacologically inactive oxidation products, one of which can be hydrolyzed by carboxylesterase to release SN-38. UGT1A1 activity is reduced in individuals with genetic polymorphisms that lead to reduced enzyme activity, such as the UGT1A1*28 polymorphism (see Precautions). SN-38 glucuronide had 1/50 to 1/100 the activity of SN-38 in cytotoxicity assays using two cell lines
in vitro. The disposition of irinotecan has not been fully elucidated in humans. The urinary excretion of irinotecan is 11% to 20%; SN-38, <1%; and SN-38 glucuronide, 3%. The cumulative biliary and urinary excretion of irinotecan and its metabolites (SN-38 and SN-38 glucuronide) over a period of 48 hours following administration of irinotecan in two patients ranged from approximately 25% (100 mg/m
2) to 50% (300 mg/m
2).
Pharmacokinetics in special populations: Geriatric: The pharmacokinetics of irinotecan administered using the weekly schedule was evaluated in a study of 183 patients that was prospectively designed to investigate the effect of age on irinotecan toxicity. Results from this trial indicate that there are no differences in the pharmacokinetics of irinotecan, SN-38, and SN-38 glucuronide in patients <65 years of age compared with patients ≥65 years of age. In a study of 162 patients that was not prospectively designed to investigate the effect of age, small (less than 18%) but statistically significant differences in dose-normalized irinotecan pharmacokinetic parameters in patients <65 years of age compared to patients ≥65 years of age were observed. Although dose-normalized AUC
0-24 for SN-38 in patients ≥65 years of age was 11% higher than in patients <65 years of age, this difference was not statistically significant.
Pediatric: see Precautions.
The pharmacokinetics of irinotecan and its major metabolites in the pediatric population was investigated in clinical trials conducted in the US and Europe. Overall, results and general conclusions regarding irinotecan pharmacokinetics were comparable in the US and European studies. Any differences in the findings between these studies are probably attributable to differences in the doses investigated (20 to 200 mg/m
2 and 200 to 720 mg/m
2 in the US and European studies, respectively) and the marked inter-patient variability in values determined for the pharmacokinetic parameters of irinotecan and SN-38.
US studies: Pharmacokinetic parameters for irinotecan and SN-38 were determined in 2 pediatric solid-tumor trials at dose levels of 50 mg/m
2 (60-min infusion, n = 48) and 125 mg/m
2 (90-min infusion, n = 6). Irinotecan clearance (mean ± S.D.) was 17.3 ± 6.7 L/h/m
2 for the 50 mg/m
2 dose and 16.2 ± 4.6 L/h/m
2 for the 125 mg/m
2 dose, which is somewhat greater than in adults. Minimal accumulation of irinotecan and SN-38 was observed in children on daily dosing regimens [daily x 5 every 3 weeks or (daily x 5) x 2 weeks every 3 weeks]. A finding that dose-normalized SN-38 AUC values were comparable between adults and children was inconsistent with the increase in irinotecan clearance seen in the pediatric population and was probably reflective of the marked inter-patient variability (%CV values for SN-38 AUC were 84% to 120%). Indeed SN-38 exposure in pediatric patients was approximately 30% lower than in adults when comparison was made without regard to the variability of the data.
European studies: The pharmacokinetics of irinotecan and its major metabolites was investigated in pediatric patients with solid tumors in a phase I study at dose levels of 200 to 720 mg/m
2 (2-hour infusion, n = 77). Systemic exposure to irinotecan, SN-38, APC, and NPC was dose proportional. Pharmacokinetic parameters of irinotecan and its metabolites demonstrated marked inter-patient variability with values (mean ± S.D.) for irinotecan plasma clearance of 18 ± 8 L/h/m
2 and volume of distribution at steady state of 104 ± 84 L/m
2. Irinotecan clearance was 26% lower in adolescents than in children and dose normalized SN-38 and SN-38G exposures were 52% and 105% higher in adolescents than in children, respectively. Irinotecan clearance was higher and dose normalized values for SN-38, SN-38G and APC exposure were lower in the pediatric than in the adult population.
A population pharmacokinetic analysis of irinotecan was performed in 83 children and adolescents with relapsed or refractory rhabdomyosarcoma, primitive neuroectodermal tumor (PNET) including medulloblastoma or neuroblastoma receiving 600 mg/m
2 irinotecan as a 1-hour infusion once every 3 weeks as part of a phase II study. Mean values for irinotecan clearance and AUC demonstrated large inter- and intra-individual variability and were similar to those determined at the same dose in the European phase-I pediatric study.
Gender: The pharmacokinetics of irinotecan do not appear to be influenced by gender.
Race: The influence of race on the pharmacokinetics of irinotecan has not been evaluated.
Hepatic Insufficiency: (see Dosage & Administration) Irinotecan clearance is diminished in patients with hepatic dysfunction while relative exposure to the active metabolite SN-38 is increased. The magnitude of these effects is proportional to the degree of liver impairment as measured by elevations in serum total bilirubin and transaminase concentrations.
Renal Insufficiency: The influence of renal insufficiency on the pharmacokinetics of irinotecan has not been evaluated (see Dosage & Administration).
Pharmacogenomics: The active metabolite SN-38 is further metabolized via UGT1A1. Genetic variants of the UGT1A1 gene such as the UGT1A1*28 [(TA)7] and *6 alleles lead to reduced UGT1A1 enzyme expression or activity and decreased function to a similar extent.
Individuals who are homozygous or compound (double) heterozygous for these alleles (e.g., *28/*28, *6/*6,*6/*28) are UGT1A1 poor metabolizers and are at increased risk for severe or life-threatening neutropenia from Irinotecan Hydrochloride, USPI due to elevated systemic exposure to SN-38. The UGT1A1*6/*6 genotype should not be confused with 6/6 genotype, which is sometimes used to represent the genotype of individuals who are wild type for UGT1A1*28. Individuals who are heterozygous for either the UGT1A1*28 or *6 alleles (*1/*6, *1/*28) are UGT1A1 intermediate metabolizers and may also have an increased risk of severe or life-threatening neutropenia (see Dosage & Administration, Precautions and Pharmacokinetics as previously mentioned).
Published studies have shown that individuals with UGT1A1*28 and *6 alleles may be at an increased risk of severe diarrhea. The risk evidence appears greater in UGT1A1*28 and *6 homozygous patients and in those taking irinotecan doses >125 mg/m
2 (see Precautions).
UGT1A1*28 and *6 alleles occur at various frequencies in different populations. Approximately 20% of Black or African American, 10% of White, and 2% of East Asian individuals are homozygous for the UGT1A1*28 allele. Approximately 2-6 % of East Asian individuals are homozygous for the UGT1A1*6 allele. The UGT1A1*6 allele is uncommon in Black or African American or in White individuals. Decreased function alleles other than UGT1A1*28 and *6 may be present in certain populations.
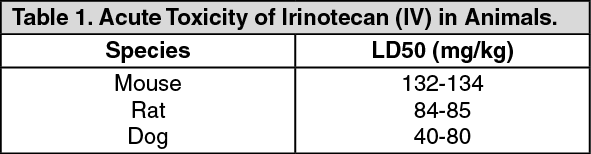
Toxicology: Preclinical safety data: The acute intravenous toxicity of irinotecan in animals is shown as follows. Lethality was observed after single intravenous irinotecan doses of approximately 111 mg/kg in mice and 73 mg/kg in rats (approximately 2.6 and 3.4 times the recommended human dose of 125 mg/m
2, respectively). Death was preceded by cyanosis, tremors, respiratory distress, and convulsions. Subacute toxicity studies show that irinotecan affects tissues with rapid cell proliferation (bone marrow, intestinal epithelia, thymus, spleen, lymph nodes, and testes). (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Carcinogenicity/Mutagenicity: Long-term carcinogenicity studies with irinotecan were not conducted. Rats were, however, administered intravenous doses of 2 mg/kg or 25 mg/kg irinotecan once per week for 13 weeks (in separate studies, the 25 mg/kg dose produced an irinotecan C
max and AUC that were about 7.0 times and 1.3 times the respective values in patients administered 125 mg/m
2) and were then allowed to recover for 91 weeks. Under these conditions, there was a significant linear trend with dose for the incidence of combined uterine horn endometrial stromal polyps and endometrial stromal sarcomas.
Neither irinotecan nor SN-38 was mutagenic in the
in vitro Ames assay. However, in the
in vitro Chinese hamster cell chromosomal aberration assay, irinotecan produced a significant increase in the incidence of chromosomal aberrations in a concentration-dependent manner. Additionally, in the
in vivo mouse micronucleus assay, a single intraperitoneal dose of irinotecan over the dosage range of 2.5 to 200 mg/kg caused a significant and dose-dependent increase in micronucleated polychromatic erythrocytes and a decrease in the reticulocyte/erythrocyte ratio in bone marrow cells.
Reproduction: No significant adverse effects on fertility and general reproductive performance were observed after intravenous administration of irinotecan in doses of up to 6 mg/kg/day to rats. However, atrophy of male reproductive organs was observed after multiple daily irinotecan doses both in rodents at 20 mg/kg (which in separate studies produced an irinotecan C
max and AUC about 5 and 1 times, respectively, the corresponding values in patients administered 125 mg/m
2) and dogs at 0.4 mg/kg (which in separate studies produced an irinotecan C
max and AUC about one-half and 1/15
th, respectively, the corresponding values in patients administered 125 mg/m
2).
Radioactivity related to
14C-irinotecan crosses the placenta of rats following intravenous administration of 10 mg/kg (which in separate studies produced an irinotecan C
max and AUC about 3 and 0.5 times, respectively, the corresponding values in patients administered 125 mg/m
2). Irinotecan was teratogenic in rats at doses greater than 1.2 mg/kg/day (which in separate studies produced an irinotecan C
max and AUC about 2/3 and 1/40
th, respectively, of the corresponding values in patients administered 125 mg/m
2) and in rabbits at 6 mg/kg/day (about one-half the recommended weekly human dose on a mg/m
2 basis). Teratogenic effects included a variety of external, visceral, and skeletal abnormalities. Irinotecan administered to rat dams for the period following organogenesis through weaning at doses of 6 mg/kg/day caused decreased learning ability and decreased female body weights in the offspring.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image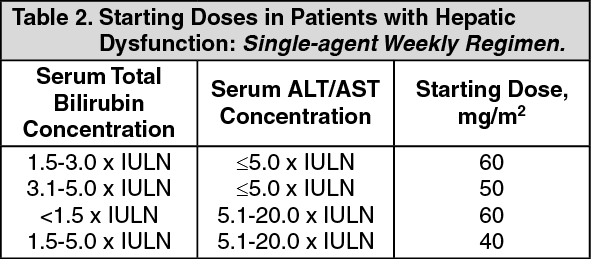 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image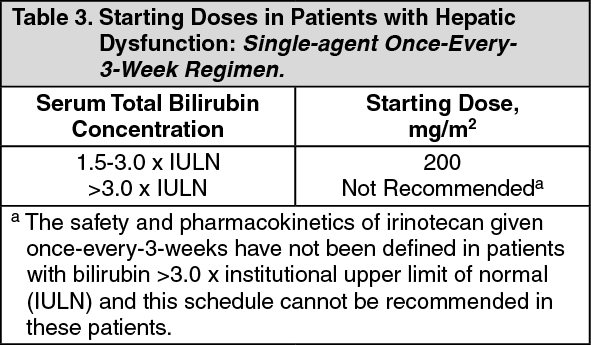 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image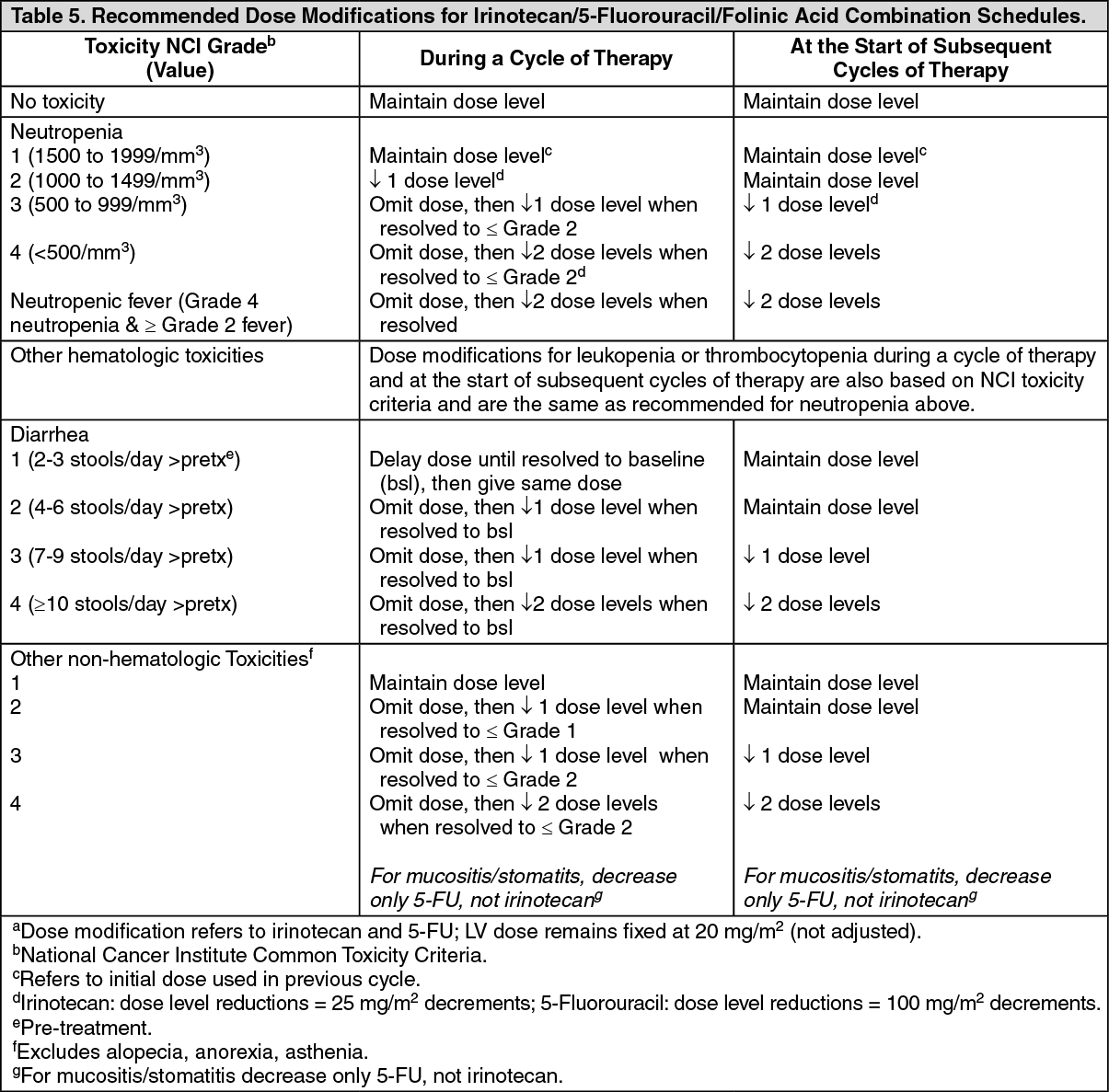 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image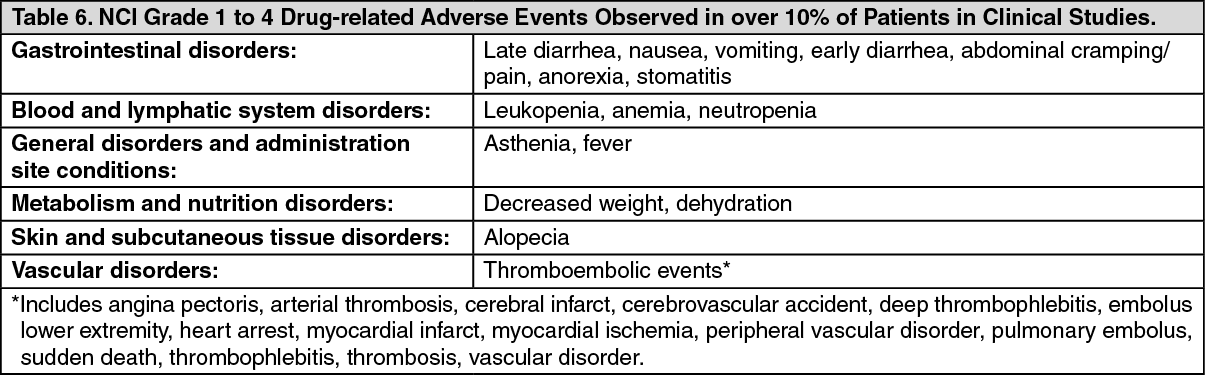 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image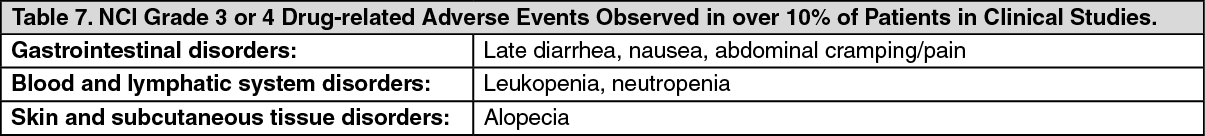 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image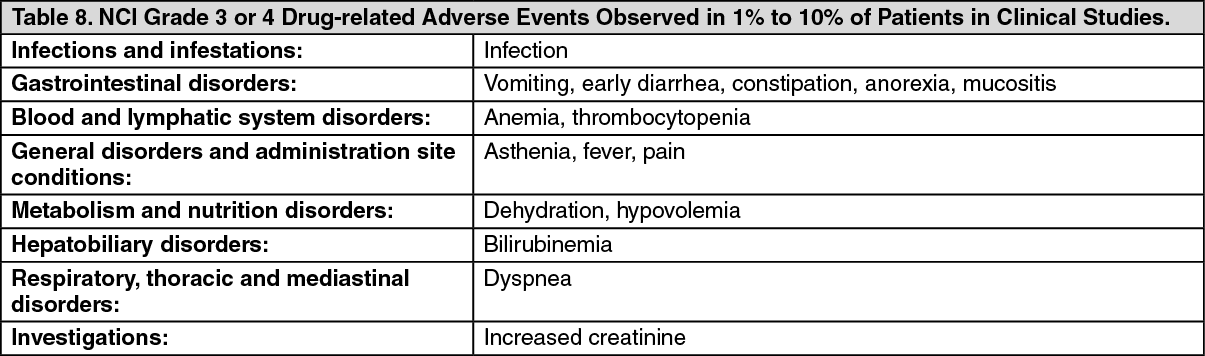 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image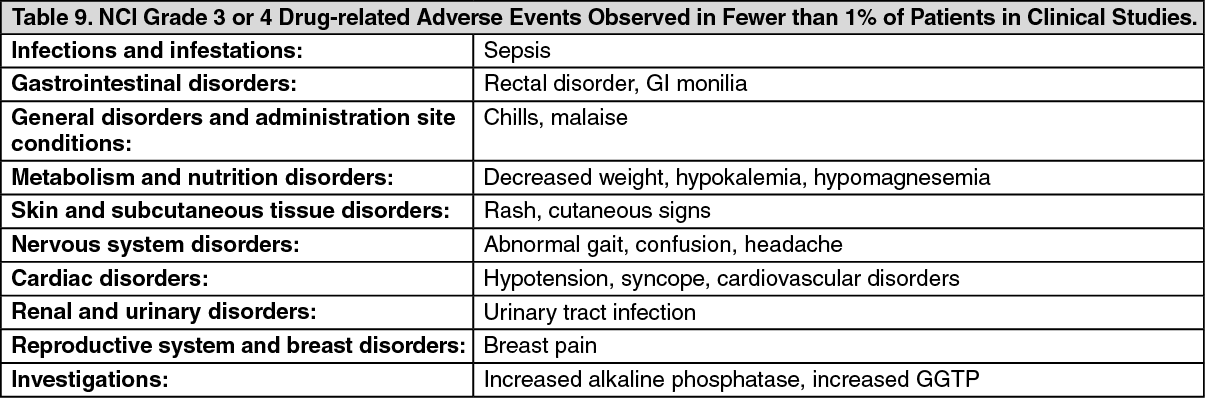 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
