Pharmacotherapeutic group: antineoplastic agent, protein kinase inhibitor.
ATC code: L01EX07.
Pharmacology: Pharmacodynamics: Mechanism of action: Cabozantinib is a small molecule that inhibits multiple receptor tyrosine kinases (RTKs) implicated in tumour growth and angiogenesis, pathologic bone remodeling, drug resistance, and metastatic progression of cancer. Cabozantinib was evaluated for its inhibitory activity against a variety of kinases and was identified as an inhibitor of MET (hepatocyte growth factor receptor protein) and VEGF (vascular endothelial growth factor) receptors. In addition, cabozantinib inhibits other tyrosine kinases including the GAS6 receptor (AXL), RET, ROS1, TYRO3, MER, the stem cell factor receptor (KIT), TRKB, Fms-like tyrosine kinase-3 (FLT3), and TIE-2.
Pharmacodynamic effects: Cabozantinib exhibited dose-related tumour growth inhibition, tumour regression, and/or inhibited metastasis in a broad range of preclinical tumour models.
Cardiac electrophysiology: An increase from baseline in corrected QT interval by Fridericia (QTcF) of 10 - 15 ms on Day 29 (but not on Day 1) following initiation of cabozantinib treatment (at a dose of 140 mg once daily) was observed in a controlled clinical trial in medullary thyroid cancer patients. This effect was not associated with a change in cardiac wave form morphology or new rhythms. No cabozantinib-treated subjects in this study had a confirmed QTcF >500 ms, nor did any cabozantinib-treated subjects in the RCC or HCC studies (at a dose of 60 mg).
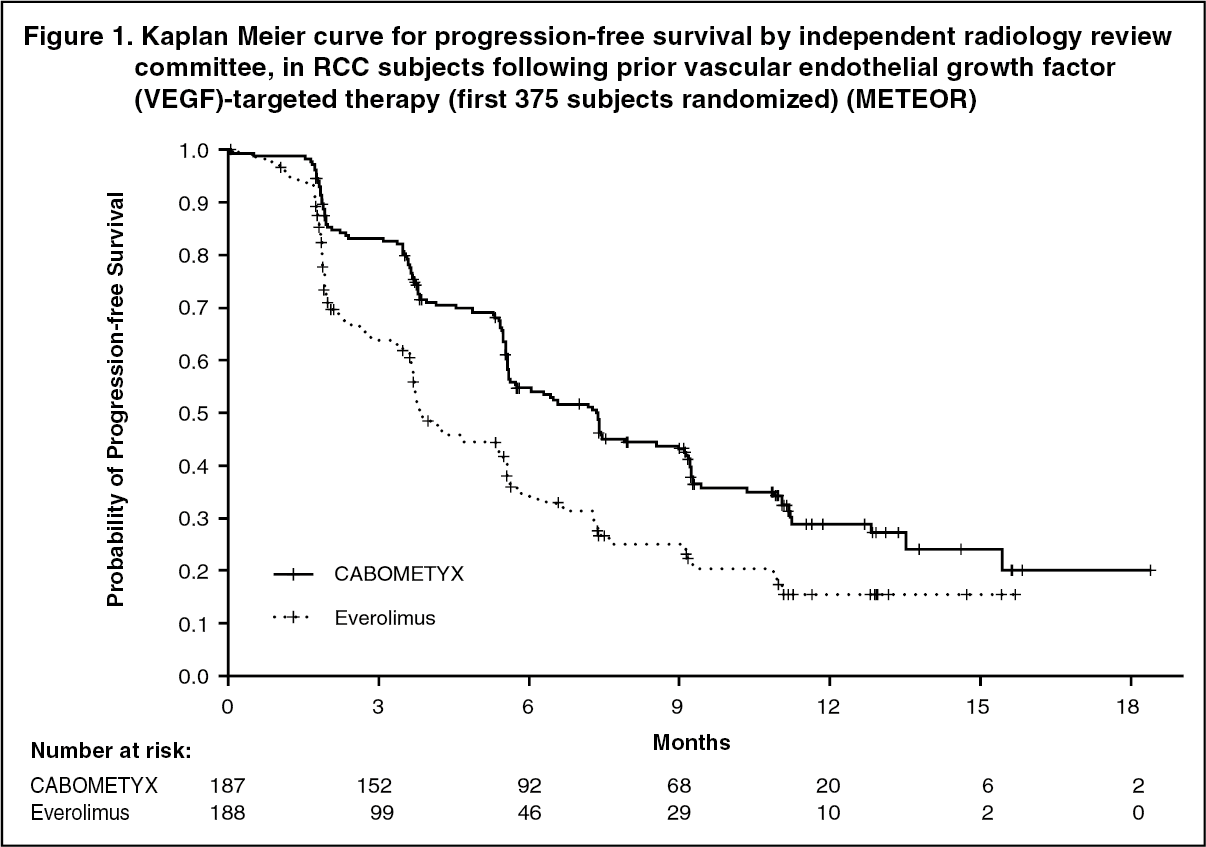
Clinical efficacy and safety: Renal cell carcinoma: Randomized study in RCC patients who have received prior vascular endothelial growth factor (VEGF)-targeted therapy (METEOR): The safety and efficacy of CABOMETYX for the treatment of renal cell carcinoma following prior vascular endothelial growth factor (VEGF)-targeted therapy were evaluated in a randomized, open-label, multicenter phase 3 study (METEOR). Patients (N=658) with advanced RCC with a clear cell component who had previously received at least 1 prior VEGF receptor tyrosine kinase inhibitor (VEGFR TKI) were randomized (1:1) to receive cabozantinib (N=330) or everolimus (N=328). Patients could have received other prior therapies, including cytokines, and antibodies targeting VEGF, the programmed death 1 (PD-1) receptor, or its ligands. Patients with treated brain metastases were allowed. Progression-free survival (PFS) was assessed by a blinded independent radiology review committee, and the primary analysis was conducted among the first 375 subjects randomized. Secondary efficacy endpoints were objective response rate (ORR) and overall survival (OS). Tumor assessments were conducted every 8 weeks for the first 12 months, then every 12 weeks thereafter.
The baseline demographic and disease characteristics were similar between the cabozantinib and everolimus arms. The majority of the patients were male (75%), with a median age of 62 years. Seventy-one percent (71%) received only one prior VEGFR TKI; 41% of patients received sunitinib as their only prior VEGFR TKI. According to the Memorial Sloan Kettering Cancer Center criteria for prognostic risk category, 46% were favorable (0 risk factors), 42% were intermediate (1 risk factor), and 13% were poor (2 or 3 risk factors). Fifty-four percent (54%) of patients had 3 or more organs with metastatic disease, including lung (63%), lymph nodes (62%), liver (29%), and bone (22%). The median duration of treatment was 7.6 months (range 0.3 - 20.5) for patients receiving cabozantinib and 4.4 months (range 0.21 - 18.9) for patients receiving everolimus.
A statistically significant improvement in PFS was demonstrated for cabozantinib compared to everolimus (Figure 1 and Table 1). A planned interim analysis of OS was conducted at the time of the PFS analysis and did not reach the interim boundary for statistical significance (202 events, HR=0.68 [0.51, 0.90], p=0.006). In a subsequent unplanned interim analysis of OS, a statistically significant improvement was demonstrated for patients randomized to cabozantinib as compared with everolimus (320 events, median of 21.4 months vs. 16.5 months; HR=0.66 [0.53, 0.83], p=0.0003; Figure 2). Comparable results for OS were observed with a follow-up analysis (descriptive) at 430 events.
Exploratory analyses of PFS and OS in the ITT population have also shown consistent results in favour of cabozantinib compared to everolimus across different subgroups according to age (<65 vs. ≥65, sex, MSKCC risk group (favourable, intermediate, poor), ECOG status (0 vs. 1), time from diagnosis to randomisation (<1 year vs. ≥1 year), tumour MET status (high vs. low vs. unknown), bone metastases (absence vs. presence), visceral metastases (absence vs. presence), visceral and bone metastases (absence vs. presence), number of prior VEGFR-TKIs (1 vs. ≥2), duration of first VEGFR-TKI (≤6 months vs. >6 months).
Objective response rate findings are summarized in Table 2. (See Figures 1, 2 and Tables 1, 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Randomized study in treatment-naïve renal cell carcinoma patients (CABOSUN): The safety and efficacy of CABOMETYX for the treatment of treatment-naïve renal cell carcinoma were evaluated in a randomized, open-label, multicenter study (CABOSUN). Patients (N=157) with previously untreated, locally advanced or metastatic RCC with a clear cell component were randomized (1:1) to receive cabozantinib (N=79) or sunitinib (N=78). Patients had to have intermediate or poor risk disease as defined by the International Metastatic RCC Database Consortium (IMDC) risk group categories. Patients were stratified by IMDC risk group and presence of bone metastases (yes/no). Approximately 75% of patients had a nephrectomy prior to onset of treatment
For intermediate risk disease, one or two of the following risk factors were met, while for poor risk, three or more factors were met: time from diagnosis of RCC to systemic treatment <1 year, Hgb < LLN, corrected calcium > ULN, KPS <80%, neutrophil count > ULN and platelet count > ULN.
The primary endpoint was PFS. Secondary efficacy endpoints were objective response rate (ORR) and overall survival (OS). Tumor assessments were conducted every 12 weeks.
The baseline demographic and disease characteristics were similar between the cabozantinib and sunitinib arms. The majority of the patients were male (78%) with a median age of 62 years. Patient distribution by IMDC risk groups was 81% intermediate (1-2 risk factors) and 19% poor (≥3 risk factors). Most patients (87%) had ECOG performance status of 0 or 1; 13% had an ECOG performance status of 2. Thirty-six percent (36%) of patients had bone metastases.
A statistically significant improvement in PFS as retrospectively assessed by a blinded Independent Radiology Committee (IRC) was demonstrated for cabozantinib compared to sunitinib (Figure 3 and Table 3). The results from the investigator determined analysis and IRC-determined analysis of PFS were consistent.
Patients with both positive and negative MET status showed a favourable effect with cabozantinib compared to sunitinib, with greater activity in patients with a positive MET status compared to patients with a negative MET status (HR=0.32 (0.16, 0.63) vs 0.67 (0.37, 1.23)) respectively.
Cabozantinib treatment was associated with a trend for longer survival compared to sunitinib (Table 3). The study was not powered for the OS analysis and the data are immature.
Objective response rate (ORR) findings are summarized in Table 3. (See Figure 3 and Table 3.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Randomised phase 3 study of cabozantinib in combination with nivolumab vs. sunitinib (CA2099ER): The safety and efficacy of cabozantinib 40 mg orally daily in combination with nivolumab 240 mg intravenously every 2 weeks for the first-line treatment of advanced/metastatic RCC was evaluated in a phase 3, randomised, open label study (CA2099ER). The study included patients (18 years or older) with advanced or metastatic RCC with a clear cell component, Karnofsky Performance Status (KPS) ≥70%, and measurable disease as per RECIST v1.1 were included regardless of their PD-L1 status or IMDC risk group. The study excluded patients with autoimmune disease or other medical conditions requiring systemic immunosuppression, patients who had prior treatment with an anti-PD-1, anti PD-L1, anti-PD-L2, anti-CD137, or anti-CTLA-4 antibody, poorly controlled hypertension despite antihypertensive therapy, active brain metastases and uncontrolled adrenal insufficiency. Patients were stratified by IMDC prognostic score, PD-L1 tumour expression, and region.
A total of 651 patients were randomised to receive either cabozantinib 40 mg once daily orally in combination with nivolumab 240 mg (n=323) administered intravenously every 2 weeks or sunitinib (n = 328) 50 mg daily, administered orally for 4 weeks followed by 2 weeks off. Treatment continued until disease progression or unacceptable toxicity with nivolumab administration up to 24 months. Treatment beyond initial Investigator-assessed RECIST version 1.1-defined progression was permitted if the patient had a clinical benefit and was tolerating study drug, as determined by investigator. First tumour assessment post-baseline was performed at 12 weeks (± 7 days) following randomisation. Subsequent tumour assessments occurred at every 6 weeks (± 7 days) until Week 60, then every 12 weeks (± 14 days) until radiographic progression, confirmed by the Blinded Independent Central review (BICR). The primary efficacy outcome measure was PFS as determined by a BICR. Additional efficacy measures included OS and ORR as key secondary endpoints.
Baseline characteristics were generally balanced between the two groups. The median age was 61 years (range: 28-90) with 38.4% ≥65 years of age and 9.5% ≥75 years of age. The majority of patients were male (73.9%) and white (81.9%). Eight percent of patients were Asian, 23.2% and 76.5% of patients had a baseline KPS of 70 to 80% and 90 to 100%, respectively. Patient distribution by IMDC risk categories was 22.6% favourable, 57.6% intermediate, and 19.7% poor. For tumour PD-L1 expression, 72.5% of patients had PD-L1 expression <1% or indeterminate and 24.9% of patients had PD-L1 expression ≥1%. 11.5% of patients had tumours with sarcomatoid features. The median duration of treatment was 14.26 months (range: 0.2-27.3 months) in cabozantinib with nivolumab-treated patients and was 9.23 months (range: 0.8-27.6 months) in sunitinib-treated patients.
The study demonstrated a statistically significant benefit in PFS, OS, and ORR for patients randomised to cabozantinib in combination with nivolumab as compared to sunitinib.
Efficacy results from the primary analysis (minimum follow-up 10.6 months; median follow-up 18.1 months) are shown in Table 4. (See Table 4.)
Click on icon to see table/diagram/image
The primary analysis of PFS included censoring for new anti-cancer treatment (Table 4). Results for PFS with and without censoring for new anti-cancer treatment were consistent.
PFS benefit was observed in the cabozantinib in combination with nivolumab arm vs. sunitinib regardless of the (IMDC) risk category. Median PFS for the favourable risk group was not reached for cabozantinib in combination with nivolumab, and was 12.81 months in the sunitinib arm (HR = 0.60; 95% CI: 0.37, 0.98). Median PFS for the intermediate risk group was 17.71 months for cabozantinib in combination with nivolumab and was 8.38 months in the sunitinib arm (HR = 0.54; 95% CI: 0.41, 0.73). Median PFS for the poor risk group was 12.29 months for cabozantinib in combination with nivolumab and was 4.21 months in the sunitinib arm (HR = 0.36; 95% CI: 0.23, 0.58).
An updated PFS and OS analysis were performed when all patients had a minimum follow-up of 16 months and a median follow-up of 23.5 months (see Figures 4 and 5). The PFS hazard ratio was 0.52 (95% CI: 0.43; 0.64). The OS hazard ratio was 0.66 (95% CI: 0.50; 0.87). Updated efficacy data (PFS and OS) in subgroups for the IMDC risk categories and PD-L1 expression levels confirmed the original results. With the updated analysis, median PFS is reached for the favourable risk group. (See Figures 4 and 5.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Hepatocellular carcinoma: Controlled study in patients who have received sorafenib (CELESTIAL): The safety and efficacy of CABOMETYX were evaluated in a randomized, double-blind, placebo-controlled Phase 3 study (CELESTIAL). Patients (N=707) with HCC not amenable to curative treatment and who had previously received sorafenib for advanced disease were randomized (2:1) to receive cabozantinib (N=470) or placebo (N=237). Patients could have received one other prior systemic therapy for advanced disease in addition to sorafenib. Randomization was stratified by aetiology of disease (HBV [with or without HCV], HCV [without HBV], or other), geographic region (Asia, other regions) and by presence of extrahepatic spread of disease and/or macrovascular invasions (Yes, No).
The primary efficacy endpoint was overall survival (OS). Secondary efficacy endpoints were progression-free survival (PFS) and objective response rate (ORR), as assessed by the Investigator using Response Evaluation Criteria in Solid Tumours (RECIST) 1.1. Tumour assessments were conducted every 8 weeks. Subjects continued blinded study treatment after radiological disease progression whilst they experienced clinical benefit or until the need for subsequent systemic or liver-directed local anticancer therapy. Crossover from placebo to cabozantinib was not allowed during the blinded treatment phase.
The baseline demographic and disease characteristics were similar between the cabozantinib and placebo arms and are shown below for all 707 randomised patients. The majority of patients (82%) were male: the median age was 64 years. The majority of patients (56%) were Caucasian and 34% of patients were Asian. Fifty three percent (53%) of patients had ECOG performance status (PS) 0 and 47% had ECOG PS 1. Almost all patients (99%) were Child Pugh A and 1% were Child Pugh B. Aetiology for HCC included 38% hepatitis B virus (HBV), 21% hepatitis C virus (HCV), 40% other (neither HBV nor HCV). Seventy-eight (78%) had macroscopic vascular invasion and/ or extra-hepatic tumour spread, 41% had alfa -fetoprotein (AFP) levels ≥400 μg/L, 44% had been treated by loco-regional transarterial embolisation or chemoinfusion procedures, 37% had radiotherapy prior to cabozantinib treatment. Median duration of sorafenib treatment was 5.32 months. Seventy-two percent (72%) of patients had received 1and 28% had received 2 prior systemic therapy regimens for advanced disease. A statistically significant improvement in OS was demonstrated for cabozantinib compared to placebo (Table 5 and Figure 6).
PFS and ORR findings are summarized in Table 5. (See Table 5, Figures 6 and 7.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The incidence of systemic non-radiation and local liver-directed systemic non-protocol anticancer therapy (NPACT) was 26% in the cabozantinib arm and 33% in the placebo arm. Subjects receiving these therapies had to discontinue study treatment. An exploratory OS analysis censoring for the use of NPACT supported the primary analysis: the HR, adjusted for stratification factors (per IxRS), was 0.66 (95% CI: 0.52, 0.84; stratified logrank p-value = 0.0005). The Kaplan-Meier estimates for median duration of OS were 11.1 months in the cabozantinib arm versus 6.9 months in the placebo arm, an estimated 4.2-month difference in the medians.
Non-disease specific quality of life (QoL) was assessed using the EuroQoL EQ-5D-5L. A negative effect of cabozantinib versus placebo on the EQ-5D utility index score was observed during the first weeks of treatment. Only limited QoL data are available after this period.
Differentiated thyroid carcinoma (DTC): Placebo-controlled study in adult patients who have received prior systemic therapy and are refractory or not eligible to radioactive iodine (COSMIC-311): The safety and efficacy of CABOMETYX was evaluated in COSMIC-311, a randomised (2:1), double-blind, placebo-controlled, multicenter trial in adult patients with locally advanced or metastatic disease with differentiated thyroid cancer that had progressed following up to two prior VEGFR-targeting therapy (including, but not limited to, lenvatinib or sorafenib) and were radioactive iodine-refractory or not eligible. Patients with measurable disease and documented radiographic progression per RECIST 1.1 per the Investigator, during or following treatment with VEGFR-targeting TKI, were randomised (N=258) to receive cabozantinib 60 mg orally once daily (N=170) or placebo (N=88).
Randomisation was stratified by prior receipt of lenvatinib (yes vs. no) and age (≤65 years vs. >65 years). Eligible patients randomised to placebo were allowed to cross-over to cabozantinib upon confirmation of progressive disease by blinded independent radiology review committee (BIRC). Subjects continued blinded study treatment as long as they experienced clinical benefit or until there was unacceptable toxicity. The primary efficacy outcome measures were progression-free survival (PFS) in the ITT population, and objective response rate (ORR) in the first 100 randomised patients, as assessed by BIRC per RECIST 1.1. Tumour assessments were conducted every 8 weeks after randomisation during the first 12 months on study, then every 12 weeks thereafter. Overall survival (OS) was an additional endpoint.
The primary analysis of PFS included 187 randomised patients, 125 to cabozantinib and 62 to placebo. Baseline demographics and disease characteristics were generally balanced for both treatment groups. The median age was 66 years (range 32 to 85 years), 51% being ≥65 years of age, 13% being ≥75 years of age. The majority of patients were white (70%), 18% of patients were Asian and 55% were female. Histologically, 55% had a confirmed diagnosis of papillary thyroid carcinoma, 48% had follicular thyroid carcinoma including 17% patients with Hürthle cell thyroid cancer. Metastases were present in 95% of the patients: lungs in 68%, lymph nodes in 67%, bone in 29%, pleura in 18% and liver in 15%. Five patients had not received prior RAI due to ineligibility, 63% had received prior lenvatinib, 60% had received prior sorafenib and 23% had received both sorafenib and lenvatinib. Baseline ECOG performance status was 0 (48%) or 1 (52%).
The median duration of treatment was 4.4 months in the cabozantinib arm and 2.3 months in the placebo arm.
The results of the primary analysis (with a cut-off date of 19 August 2020 and median follow up 6.2 months for the PFS), and the updated analysis (with a cut-off date of 08 February 2021 and median follow-up 10.1 months for the PFS) are presented in Table 6. The trial did not demonstrate a statistically significant improvement in ORR for patients randomised to cabozantinib (n=67) compared with placebo (n=33): 15% vs. 0%. The trial demonstrated a statistically significant improvement in PFS (median follow up 6.2 months) for patients randomised to cabozantinib (n=125) compared with placebo (n=62).
An updated analysis of PFS and OS (median follow up 10.1 months) was performed including 258 randomised patients, 170 to cabozantinib and 88 to placebo.
The overall survival analysis was confounded as placebo-treated subjects with confirmed disease progression had the option to cross over to cabozantinib. (See Table 6 and Figure 8.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Paediatric population:
The European Medicines Agency has deferred the obligation to submit the results of studies with CABOMETYX in one or more subsets of the paediatric population in treatment of solid malignant tumours (see Dosage & Administration for information on paediatric use).
Pharmacokinetics: Absorption: Following oral administration of cabozantinib, peak cabozantinib plasma concentrations are reached at 3 to 4 hours post-dose. Plasma-concentration time profiles show a second absorption peak approximately 24 hours after administration, which suggests that cabozantinib may undergo enterohepatic recirculation.
Repeat daily dosing of cabozantinib at 140 mg for 19 days resulted in an approximately a 4- to 5-fold mean cabozantinib accumulation (based on AUC) compared to a single dose administration; steady state is achieved by approximately Day 15.
A high-fat meal moderately increased C
max and AUC values (41% and 57%, respectively) relative to fasted conditions in healthy volunteers administered a single 140 mg oral cabozantinib dose. There is no information on the precise food-effect when taken 1 hour after administration of cabozantinib.
Bioequivalence could not be demonstrated between the cabozantinib capsule and tablet formulations following a single 140 mg dose in healthy subjects. A 19% increase in the C
max of the tablet formulation compared to the capsule formulation was observed. A less than 10% difference in the AUC was observed between cabozantinib tablet and capsule formulations.
Distribution: Cabozantinib is highly protein bound
in vitro in human plasma (≥99.7%). Based on the population-pharmacokinetic (PK) model, the volume of distribution of the central compartment (Vc/F) was estimated to be 212 L.
Biotransformation: Cabozantinib was metabolized
in vivo. Four metabolites were present in plasma at exposures (AUC) greater than 10% of parent: XL184-N-oxide, XL184 amide cleavage product, XL184 monohydroxy sulfate, and 6-desmethyl amide cleavage product sulfate. Two non-conjugated metabolites (XL184-N-oxide and XL184 amide cleavage product), which possess <1% of the on-target kinase inhibition potency of parent cabozantinib, each represent <10% of total drug-related plasma exposure.
Cabozantinib is a substrate for CYP3A4 metabolism
in vitro, as a neutralizing antibody to CYP3A4 inhibited formation of metabolite XL184 N-oxide by >80% in a NADPH-catalyzed human liver microsomal (HLM) incubation; in contrast, neutralizing antibodies to CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C19, CYP2D6 and CYP2E1 had no effect on cabozantinib metabolite formation. A neutralizing antibody to CYP2C9 showed a minimal effect on cabozantinib metabolite formation (i.e., a <20% reduction).
Elimination: In a population PK analysis of cabozantinib using data collected from 1883 patients and 140 healthy volunteers following oral administration of a range of doses from 20 to 140 mg, the plasma terminal half-life of cabozantinib is approximately 110 hours. Mean clearance (CL/F) at steady-state was estimated to be 2.48 L/hr. Within a 48-day collection period after a single dose of
14C-cabozantinib in healthy volunteers, approximately 81% of the total administered radioactivity was recovered with 54% in faeces and 27% in urine.
Pharmacokinetics in special patient populations: Renal impairment: In a renal impairment study conducted with a single 60 mg dose of cabozantinib, the ratios of geometric LS mean for total plasma cabozantinib, C
max and AUC
0-inf were 19% and 30% higher, for subjects with mild renal impairment (90% CI for C
max 91.60% to 155.51%; AUC
0-inf 98.79% to 171.26%) and 2% and 6-7% higher (90% CI for C
max 78.64% to 133.52%; AUC
0-inf 79.61% to 140.11%), for subjects with moderate renal impairment compared to subjects with normal renal function. The geometric LS means for unbound plasma cabozantinib AUC
0-inf was 0.2% higher for subjects with mild renal impairment (90% CI 55.9% to 180%) and 17% higher (90% CI 65.1% to 209.7%) for subjects with moderate renal impairment compared to subjects with normal renal function. Subjects with severe renal impairment have not been studied.
Hepatic impairment: Based on an integrated population pharmacokinetic analysis of cabozantinib in healthy subjects and cancer patients (including HCC), no clinically significant difference in the mean cabozantinib plasma exposure was observed amongst subjects with normal liver function (n=1425) and mild hepatic impairment (n=558). There is limited data in patients with moderate hepatic impairment (n=15) as per NCI-ODWG (National Cancer Institute - Organ Dysfunction working Group) criteria. The pharmacokinetics of cabozantinib was not evaluated in patients with severe hepatic impairment.
Race: A population PK analysis did not identify clinically relevant differences in PK of cabozantinib based on race.
Paediatrics: Data obtained from simulation performed with the population pharmacokinetic model developed in healthy subjects as well as adult patients with different type of malignancies show that in adolescent patients aged 12 years and older, a dose of 40 mg of cabozantinib once daily for patients <40 kg, or a dose of 60 mg once daily in patients ≥40 kg results in a similar plasma exposure attained in adults treated with 60 mg of cabozantinib once daily (see Dosage & Administration).
Toxicology: Preclinical safety data: Adverse reactions not observed in clinical trials, but seen in animals at exposure levels similar to clinical exposure levels and with possible relevance to clinical use were as follows: In rat and dog repeat-dose toxicity studies up to 6 months duration, target organs for toxicity were GI tract, bone marrow, lymphoid tissues, kidney, adrenal and reproductive tract tissues. The no observed adverse effect level (NOAEL) for these findings were below human clinical exposure levels at intended therapeutic dose.
Cabozantinib has shown no mutagenic or clastogenic potential in a standard battery of genotoxicity assays. The carcinogenic potential of cabozantinib has been evaluated in two species: rasH2 transgenic mice and Sprague-Dawley rats. In the 2-year rat carcinogenicity study, cabozantinib-related neoplastic findings consisted of an increased incidence of benign pheochromocytoma, alone or in combination with malignant pheochromocytoma/complex malignant pheochromocytoma of the adrenal medulla in both sexes at exposures well below the intended exposure in humans. The clinical relevance of the observed neoplastic lesions in rats is uncertain, but likely to be low.
Cabozantinib was not carcinogenic in the rasH2 mouse model at a slightly higher exposure than the intended human therapeutic exposure.
Fertility studies in rats have shown reduced male and female fertility. Further, hypospermatogenesis was observed in male dogs at exposure levels below human clinical exposure levels at intended therapeutic dose.
Embryo-foetal development studies were performed in rats and rabbits. In rats, cabozantinib caused post-implantation loss, foetal oedema, cleft palate/lip, dermal aplasia and kinked or rudimentary tail. In rabbits, cabozantinib produced foetal soft tissue changes (reduced spleen size, small or missing intermediate lung lobe) and increased foetal incidence of total malformations. NOAEL for embryo-foetal toxicity and teratogenic findings were below human clinical exposure levels at intended therapeutic dose.
Juvenile rats (comparable to a >2 year-old paediatric population) administered cabozantinib showed increased WBC parameters, decreased haematopoiesis, pubescent/immature female reproductive system (without delayed vaginal opening), tooth abnormalities, reduced bone mineral content and density, liver pigmentation and lymph node lymphoid hyperplasia. Findings in uterus/ovaries and decreased haematopoiesis appeared to be transient, while effects on bone parameters and liver pigmentation were sustained. Juvenile rats (correlating to a <2-year paediatric population) showed similar treatment-related findings, with additional findings in male reproductive system (degeneration and/or atrophy of seminiferous tubules in testes, reduced luminal sperm in epididymis), and appeared to be more sensitive to cabozantinib-related toxicity at comparable dose levels.




 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image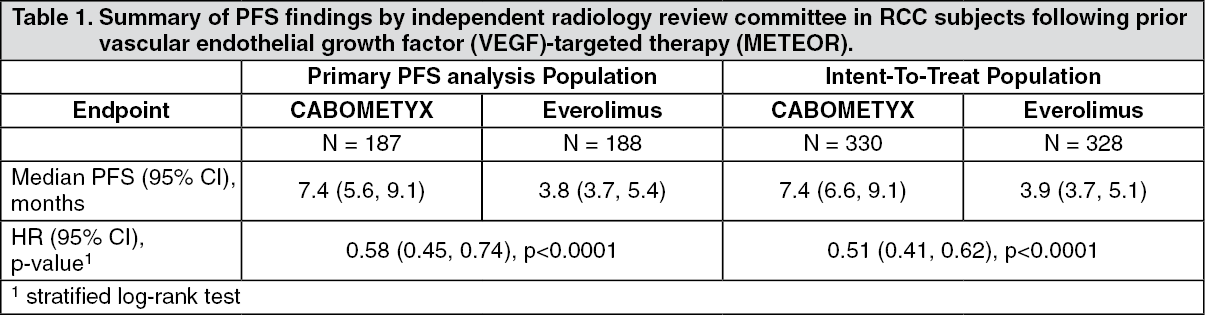 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image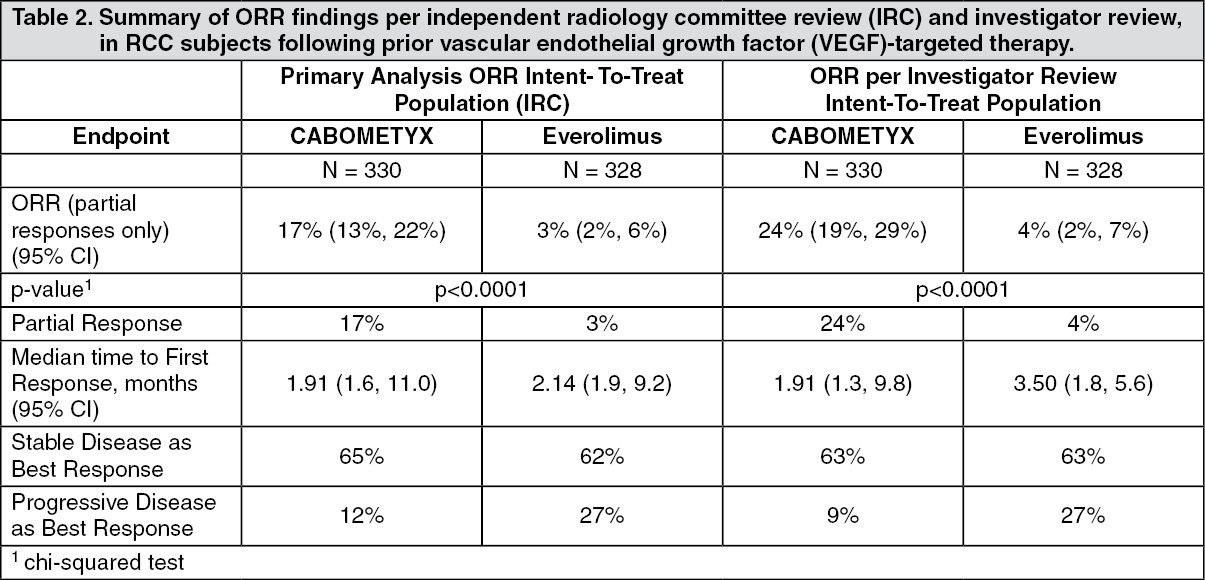 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image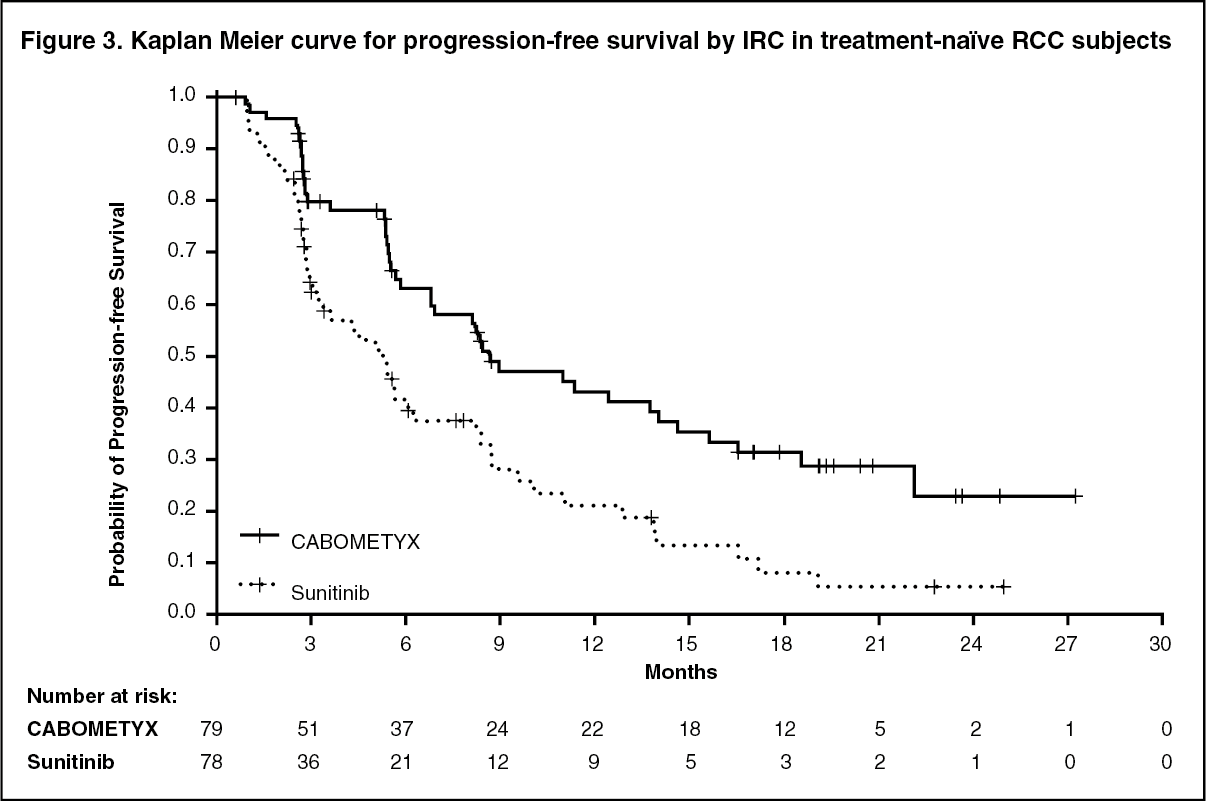 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image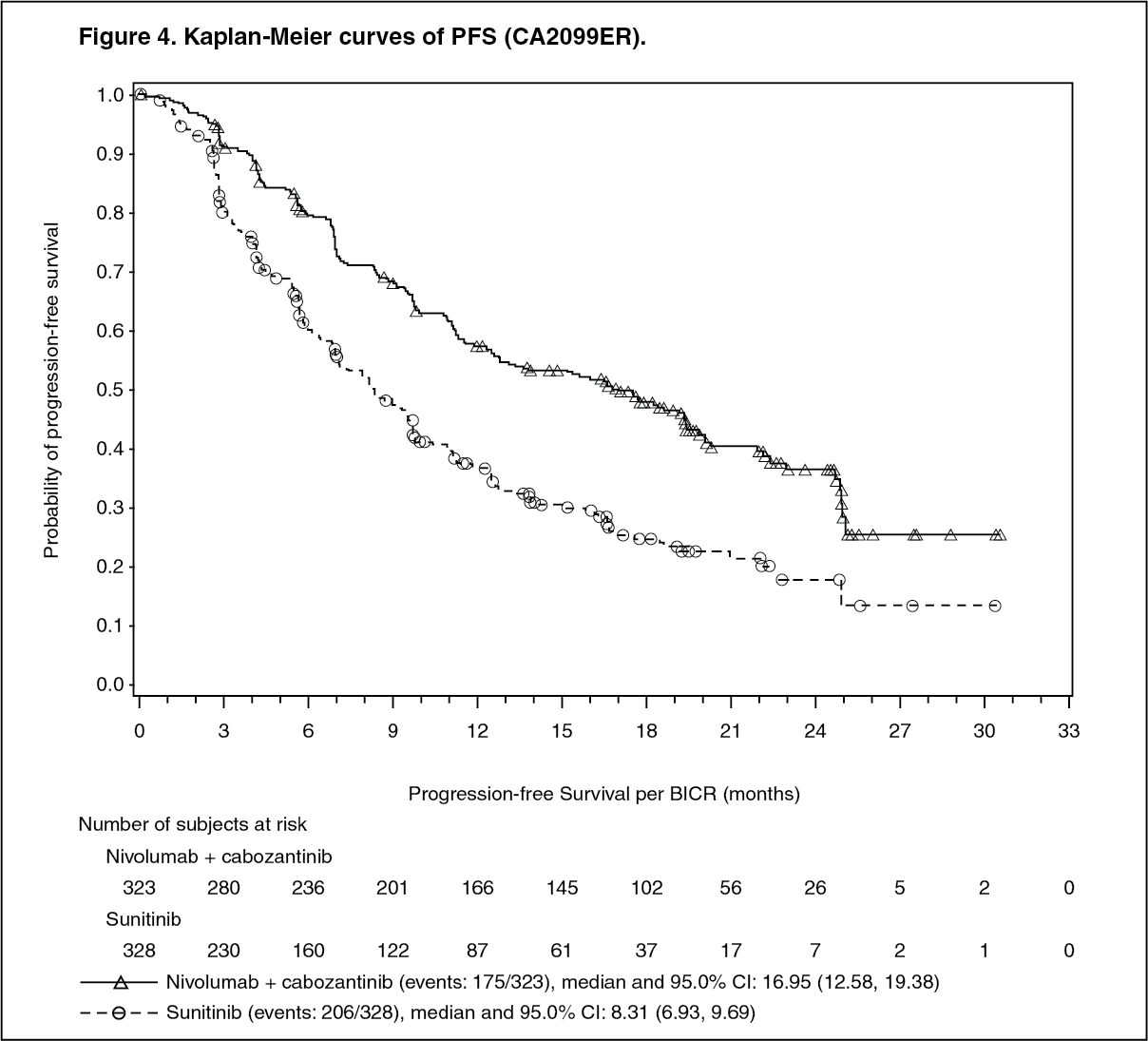 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image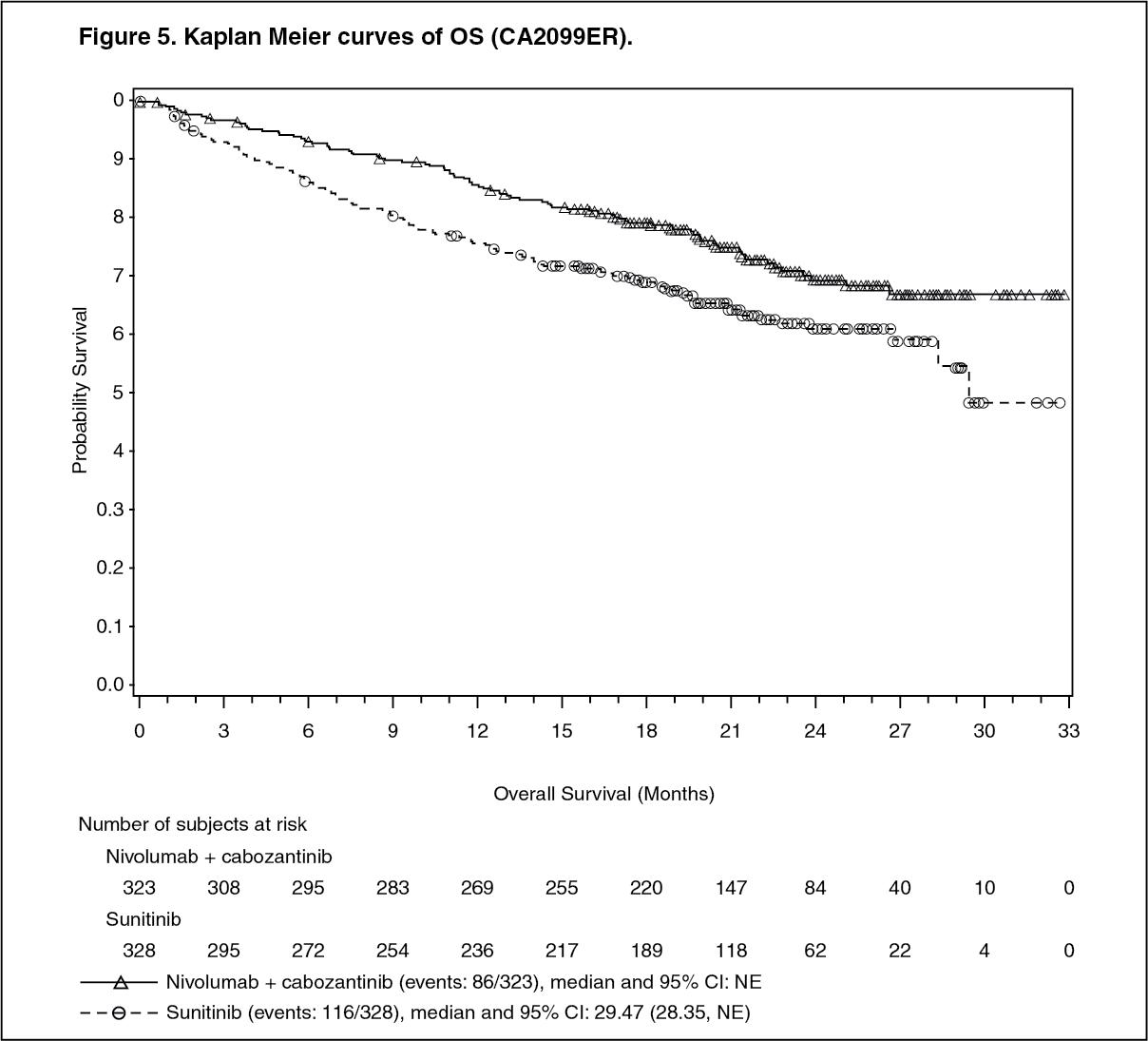 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image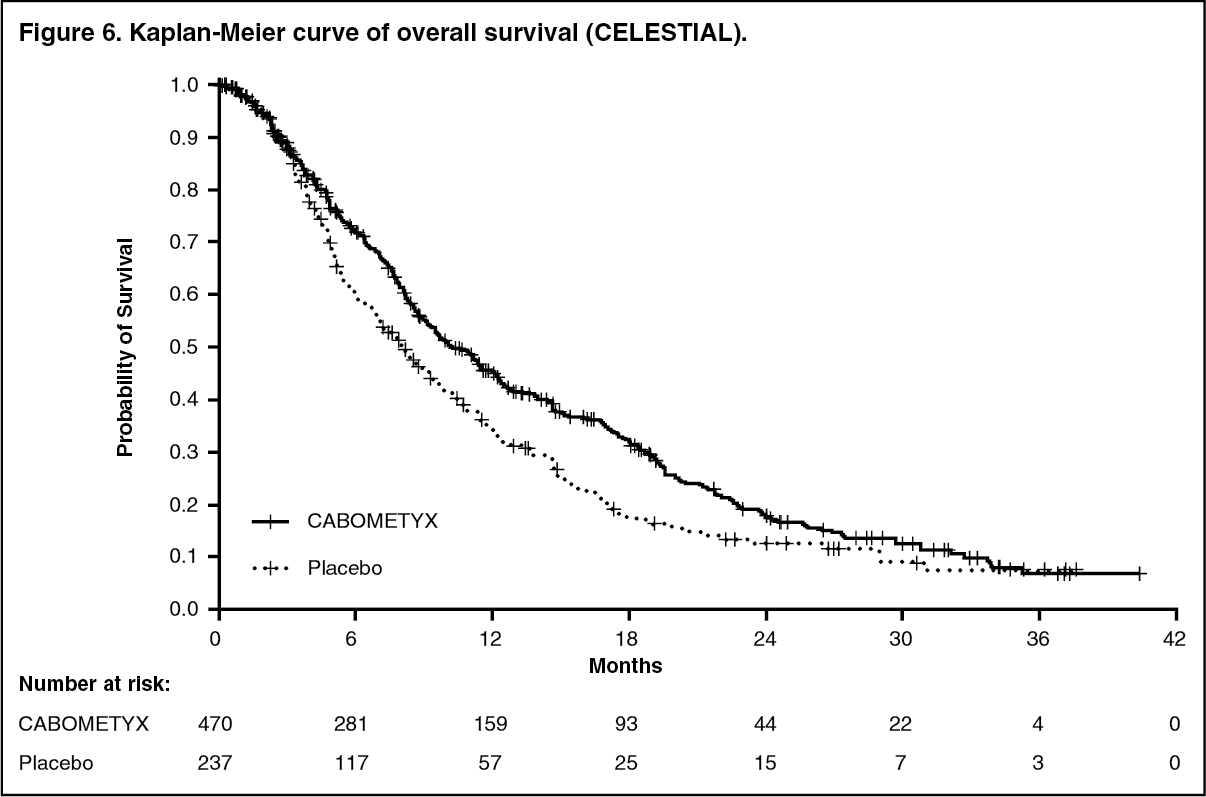 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image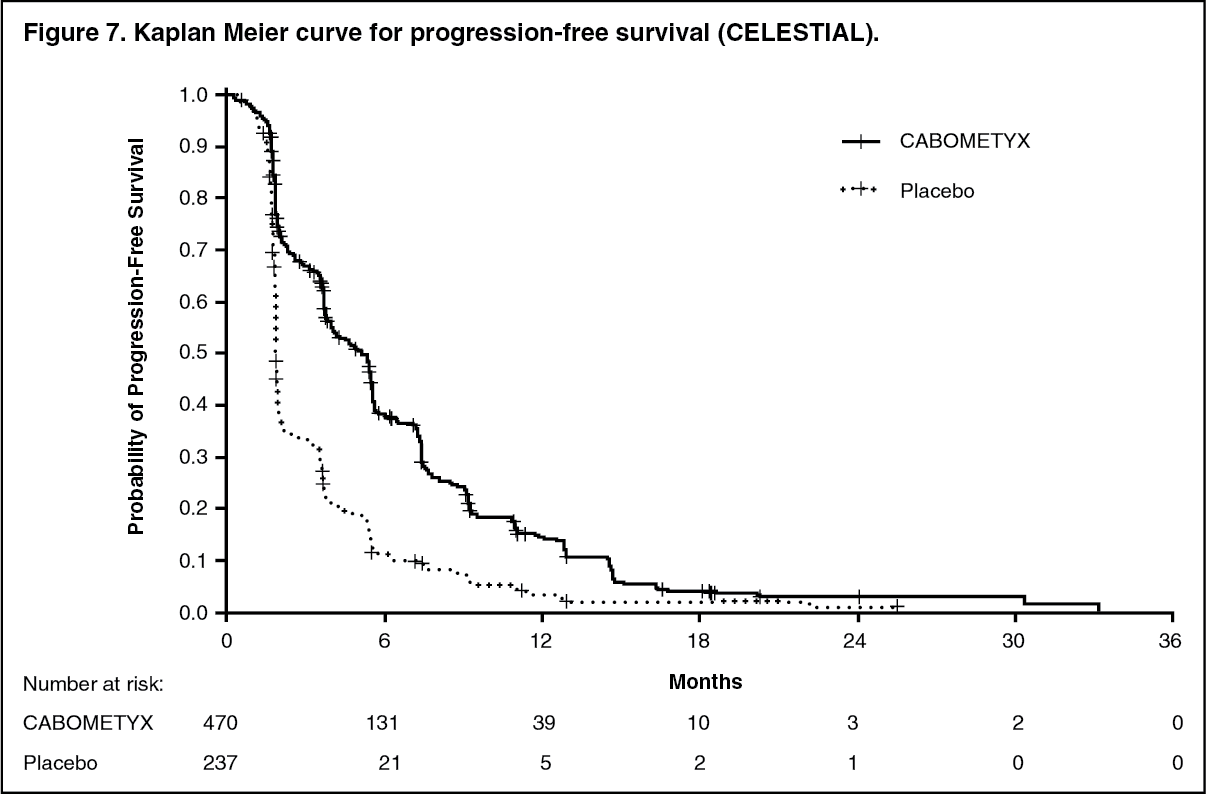 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out
