Throughout this section, adverse reactions are presented. Adverse reactions are adverse events that were considered to be reasonably associated with the use of abiraterone acetate based on the comprehensive assessment of the available adverse event information. A causal relationship with abiraterone acetate cannot be reliably established in individual cases. Further, because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
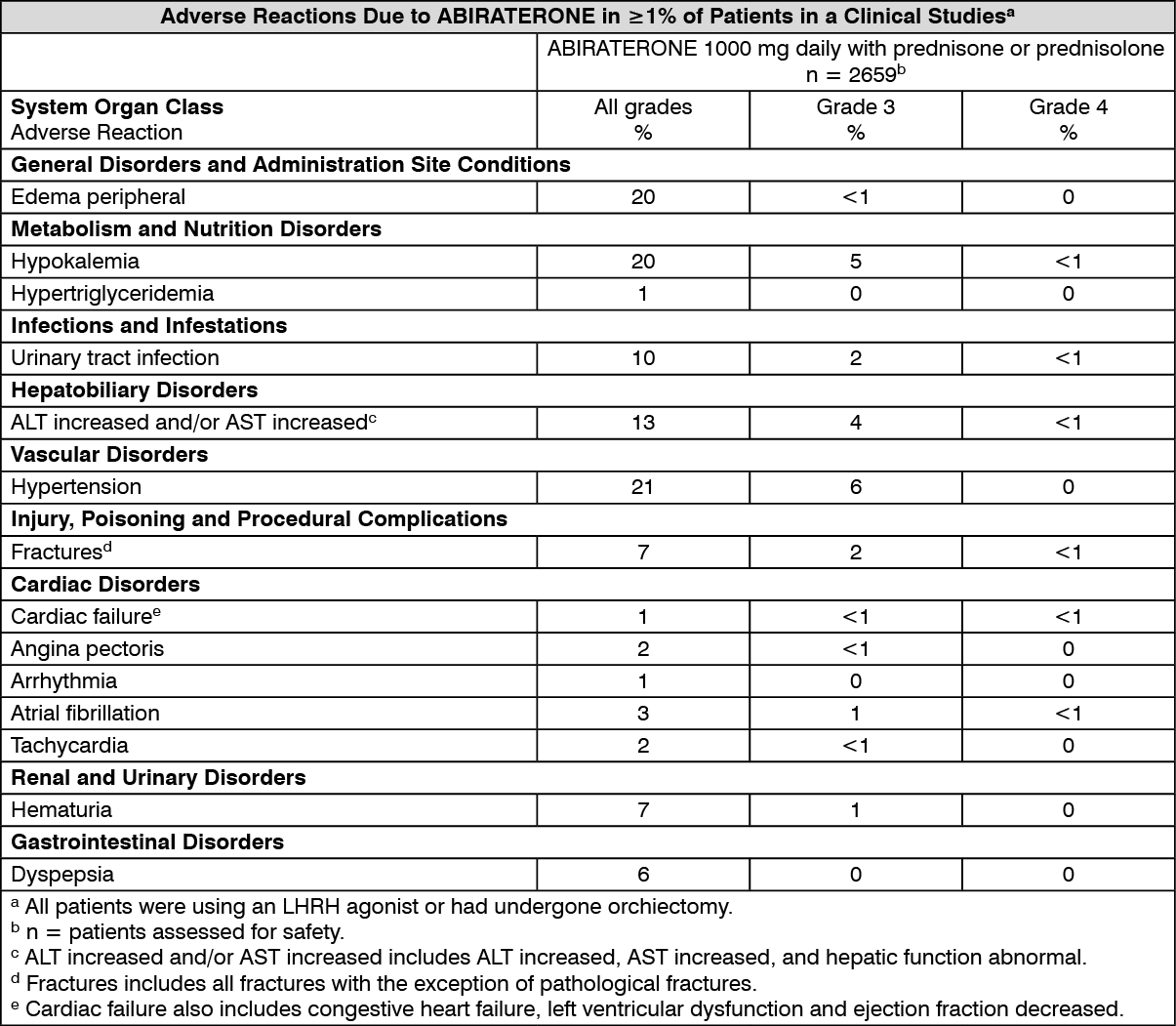
In an analysis of adverse reaction of composite Phase 3 studies with ABIRATERONE, adverse reactions that were observed in ≥ 10% of patients were hypertension, peripheral edema, hypokalemia, urinary tract infection, and aspartate aminotransferase increased and/or alanine aminotransferase increased.
ABIRATERONE may cause hypertension, hypokalemia and fluid retention as a pharmacodynamic consequence of its mechanism of action. In Phase 3 studies anticipated mineralocorticoid effects were seen more commonly in patients treated with ABIRATERONE versus patients treated with placebo: hypokalemia 18% versus 8%, hypertension 22% versus 16% and fluid retention (peripheral edema) 23% versus 17%, respectively. In patients treated with ABIRATERONE, grades 3 and 4 hypokalemia were observed in 6% and 2% of patients, grades 3 and 4 hypertension were observed in 8% and 5% of patients, and grades 3 and 4 fluid retention edema were observed in 1% and 1% of patients, respectively. Mineralocorticoid effects generally were able to be successfully managed medically. Concomitant use of a corticosteroid reduces the incidence and severity of these adverse reactions (see Hypertension, hypokalaemia, fluid retention and cardiac failure due to mineralocorticoid excess under Precautions).
In studies of patients with metastatic advanced prostate cancer who were using a LHRH agonist, or were previously treated with orchiectomy, ABIRATERONE was administered at a dose of 1000 mg daily in combination with low dose prednisone or prednisolone (5 or 10 mg daily).
Adverse reactions that occurred at a rate of ≥ 1% (all grades) are shown in table as follows: (See table.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The adverse reaction, adrenal insufficiency, occurred in the Phase 3 clinical studies at a rate of 0.3% in patients taking ABIRATERONE and at a rate of 0.1% in patients taking placebo.
Cardiovascular effects: The three Phase 3 studies excluded patients with uncontrolled hypertension, clinically significant heart disease as evidenced by myocardial infarction, arterial thrombotic events in the past 6 months, severe or unstable angina, NYHA Class III or IV heart failure (Study 301) or Class II to IV heart failure (Studies 3011 and 302) or cardiac ejection fraction measurement of < 50%. All patients enrolled (both active and placebo-treated patients) were concomitantly treated with androgen deprivation therapy, predominantly with the use of LHRH agonists, which has been associated with diabetes, myocardial infarction, cerebrovascular accident and sudden cardiac death. The incidence of cardiovascular adverse reactions in the Phase 3 studies in patients taking ABIRATERONE versus patients taking placebo were as follows: atrial fibrillation 2.6% vs. 2.0%, tachycardia 1.9% vs. 1.0%, angina pectoris 1.7% vs. 0.8%, cardiac failure 0.7% vs. 0.2% and arrhythmia 0.7% vs. 0.5%.
Hepatotoxicity: Drug associated hepatotoxicity with elevated ALT, aspartate transaminase (AST) and total bilirubin has been reported in patients treated with ABIRATERONE. Across Phase 3 clinical studies, hepatotoxicity grades 3 and 4 (e.g., ALT or AST increases of > 5X ULN or bilirubin increases > 1.5X ULN) were reported in approximately 6% of patients who received ABIRATERONE, typically during the first 3 months after starting treatment. In Study 3011, grade 3 or 4 hepatotoxicity was observed in 8.4% of patients treated with ABIRATERONE. Ten patients who received ABIRATERONE were discontinued because of hepatotoxicity; two had Grade 2 hepatotoxicity, six had Grade 3 hepatotoxicity, and two had Grade 4 hepatotoxicity. No patient died of hepatotoxicity in Study 3011. In the Phase 3 clinical studies, patients whose baseline ALT or AST was elevated were more likely to experience liver function test elevations than those beginning with normal values. When elevations of either ALT or AST > 5X ULN, or elevations in bilirubin > 3X ULN were observed, ABIRATERONE was withheld or discontinued. In two instances marked increases in liver function tests occurred (see Hepatotoxicity and Hepatic impairment under Precautions). These two patients with normal baseline hepatic function, experienced ALT or AST elevations 15 to 40X ULN and bilirubin elevations 2 to 6X ULN. Upon discontinuation of ABIRATERONE, both patients had normalisation of their liver function tests and one patient was re-treated with ABIRATERONE without recurrence of the elevations. In Study 302, grade 3 or 4 ALT or AST elevations were observed in 35 (6.5%) patients treated with ABIRATERONE. Aminotransferase elevations resolved in all but 3 patients (2 with new multiple liver metastases and 1 with AST elevation approximately 3 weeks after the last dose of ABIRATERONE). In Phase 3 clinical studies, treatment discontinuations due to ALT and AST increases or abnormal hepatic function were reported in 1.1% of patients treated with ABIRATERONE and 0.6% of patients treated with placebo; no deaths were reported due to hepatotoxicity event.
In clinical trials, the risk for hepatotoxicity was mitigated by exclusion of patients with baseline hepatitis or significant abnormalities of liver function tests. In the 3011 trial, patients with baseline ALT and AST > 2.5X ULN, bilirubin > 1.5X ULN or those with active or symptomatic viral hepatitis or chronic liver disease; ascites or bleeding disorders secondary to hepatic dysfunction were excluded. In the 301 trial, patients with baseline ALT and AST ≥ 2.5X ULN in the absence of liver metastases and > 5X ULN in the presence of liver metastases were excluded. In the 302 trial patients with liver metastases were not eligible and patients with baseline ALT and AST ≥ 2.5X ULN were excluded. Abnormal liver function tests developing in patients participating in clinical trials were vigorously managed by requiring treatment interruption and permitting re-treatment only after return of liver function tests to the patient's baseline (see Dosage & Administration). Patients with elevations of ALT or AST > 20X ULN were not re-treated. The safety of re-treatment in such patients is unknown. The mechanism for hepatotoxicity associated with ABIRATERONE is not understood.
Post-marketing experience: Adverse reactions identified during the post-marketing experience based on spontaneous reports with ABIRATERONE are described as follows. The frequencies are provided according to the following convention: Uncommon ≥ 1/1000 and < 1/100, Rare ≥ 1/10000 and < 1/1000, Very Rare < 1/10000.
System Organ Class: Respiratory, thoracic and mediastinal disorders: Rare: Allergic alveolitis.
System Organ Class: Musculoskeletal and connective tissue disorders: Uncommon: Rhabdomyolysis, Myopathy.
System Organ Class: Hepatobiliary disorders: Rare: Hepatitis fulminant, Acute hepatic failure.
System Organ Class: Cardiac disorders: Very rare: QT prolongation and Torsades de Pointes (observed in patients who developed hypokalemia or had underlying cardiovascular conditions).
System Organ Class: Immune System Disorders - Hypersensitivity.
Very rare: Anaphylactic reaction (severe allergic reactions that include, but are not limited to difficulty swallowing or breathing, swollen face, lips, tongue or throat, or an itchy rash (urticaria)).


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
