


 Sign Out
Sign Out

Antiviral activity: Remdesivir exhibited in vitro activity against a clinical isolate of SARS-CoV-2 in primary human airway epithelial cells with a 50% effective concentration (EC50) of 9.9 nM after 48 hours of treatment. The EC50 values of Remdesivir against SARS-CoV-2 in Vero cells were 137 nM at 24 hours and 750 nM at 48 hours post-treatment. The antiviral activity of Remdesivir was antagonised by chloroquine phosphate in a dose-dependent manner when the two drugs were co-incubated at clinically relevant concentrations in HEp-2 cells infected with respiratory syncytial virus (RSV). Higher Remdesivir EC50 values were observed with increasing concentrations of chloroquine phosphate. Increasing concentrations of chloroquine phosphate reduced formation of Remdesivir triphosphate in normal human bronchial epithelial cells.
Resistance: Cell culture resistance profiling of Remdesivir using the rodent CoV murine hepatitis virus identified 2 substitutions (F476L and V553L) in the viral RNA-dependent RNA polymerase at residues conserved across CoVs that conferred 5.6-fold reduced susceptibility to Remdesivir. Introduction of the corresponding substitutions (F480L and V557L) into SARS-CoV resulted in 6-fold reduced susceptibility to Remdesivir cell culture and attenuated SARS-CoV pathogenesis in a mouse model.
The cell culture development of SARS-CoV-2 resistance to Remdesivir has not been assessed to date. No clinical data are available on the development of SARS-CoV-2 resistance to Remdesivir.
Clinical Studies: NIAID ACTT-1 Trial in Subjects with Mild/Moderate and Severe COVID-19: A randomized, double-blind, placebo-controlled clinical trial evaluated Remdesivir 200 mg once daily for 1 day followed by Remdesivir 100 mg once daily for 9 days (for a total of up to 10 days of intravenously administered therapy) in hospitalized adult subjects with COVID-19 with evidence of lower respiratory tract involvement. Treatment with Remdesivir was stopped in subjects who were discharged from the hospital prior to the completion of 10 days of treatment.
The trial enrolled 1,062 subjects: 105 (9.9%) subjects with mild/moderate disease and 957 (90.1%) subjects with severe disease. A total of 285 subjects (26.8%) (n=131 received Remdesivir) were on invasive mechanical ventilation/ECMO. Subjects were randomized in a 1:1 manner, stratified by disease severity at enrollment, to receive Remdesivir (n=541) or placebo (n=521), plus standard of care.
The primary clinical endpoint was time to recovery within 29 days after randomization, defined as either discharged from the hospital or hospitalized but not requiring supplemental oxygen and no longer requiring ongoing medical care. The median time to recovery was 10 days in the Remdesivir group compared to 15 days in the placebo group recovery rate ratio, 1.29; [95% CI 1.12 to 1.49]; p<0.001). Overall, the odds of improvement in the ordinal scale were higher in the Remdesivir group at Day 15 when compared to the placebo group (odds ratio, 1.6; [95% CI, 1.3 to 1.9]; p<0.001). In a preliminary analysis conducted after 607 recoveries, 14-day mortality was 7.1% for the Remdesivir group versus 11.9% for the placebo group (hazard ratio, 0. 70 [95% CI 0.47, 1.04]; p=0.07).
Among subjects with mild/moderate disease at enrollment (n=105), the median time to recovery was 5 days in both the Remdesivir and placebo groups (recovery rate ratio, 1.22; [95% CI 0.82 to 1.81]); the odds of improvement in the ordinal scale in the Remdesivir group at Day 15 when compared to the placebo group wereas follows: odds ratio, 1.46; [95% CI, 0.71 to 2.97].
Among subjects with severe disease at enrollment (n=957), the median time to recovery was 11 days in the Remdesivir group compared to 18 days in the placebo group (recovery rate ratio, 1.31; [95% CI, 1.12 to 1.52]; p<0.001); the odds of improvement in the ordinal scale in the Remdesivir group at Day 15 when compared to the placebo group were as follows: odds ratio, 1.56; [95% CI, 1.24 to 1.95].
Study GS-US-540-5773 in Subjects with Severe COVID-19: A randomized, open-label multi-center clinical trial (Study GS-US-540-5773) of hospitalized subjects at least 12 years of age with confirmed SARS-CoV-2 infection, oxygen saturation of ≤94% on room air, and radiological evidence of pneumonia compared 200 subjects who received IV Remdesivir for 5 days with 197 subjects who received IV Remdesivir for 10 days. Patients on mechanical ventilation at screening were excluded. All subjects received 200 mg of Remdesivir on Day 1 and 100 mg once daily on subsequent days, plus standard of care. Treatment with Remdesivir was stopped in subjects who were discharged from the hospital prior to completion of their protocol-defined duration of treatment.
The primary endpoint was clinical status on Day 14 assessed on a 7-point ordinal scale consisting of the following categories: 1, death; 2, hospitalized, receiving invasive mechanical ventilation or ECMO; 3, hospitalized, receiving noninvasive ventilation or high-flow oxygen devices; 4, hospitalized, requiring low-flow supplemental oxygen; 5, hospitalized, not requiring supplemental oxygen but receiving ongoing medical care (related or not related to COVID-19); 6, hospitalized, requiring neither supplemental oxygen nor ongoing medical care (other than that specified in the protocol for Remdesivir administration); and 7, not hospitalized. After adjusting for between-group differences at baseline, patients receiving a 10-day course of Remdesivir had similar clinical status at Day 14 as those receiving a 5-day course (odds ratio for improvement, 0.75; [95% CI 0.51 to 1.12]). 28-day mortality was 11.5% and 14.2% in the 5- and 10-day treatment groups, respectively.
Study GS-US-540-5774 in Subjects with Moderate COVID-19: A randomized, open-label multi-center clinical trial (Study GS-US-540-5774) of hospitalized subjects at least 12 years of age with confirmed SARS-CoV-2 infection and radiological evidence of pneumonia without an oxygen requirement during screening compared treatment with Remdesivir for 5 days (n=191) and treatment with Remdesivir for 10 days (n=193) with standard of care (SOC) (n=200). Subjects treated with Remdesivir received 200 mg on Day 1 and 100 mg once daily on subsequent days. Treatment with Remdesivir was stopped in subjects who were discharged from the hospital prior to completion of their protocol-defined duration of treatment.
The primary endpoint was clinical status on Day 11 assessed on a 7-point ordinal scale consisting of the following categories: 1, death; 2, hospitalized, receiving invasive mechanical ventilation or ECMO; 3, hospitalized, receiving noninvasive ventilation or high-flow oxygen devices; 4, hospitalized, requiring low-flow supplemental oxygen; 5, hospitalized, not requiring supplemental oxygen but receiving ongoing medical care (related or not related to COVID-19); 6, hospitalized, requiring neither supplemental oxygen nor ongoing medical care (other than that specified in the protocol for Remdesivir administration); and 7, not hospitalized. Baseline clinical status, oxygen support status, and median duration of symptoms and hospitalization prior to first dose of Remdesivir were similar across treatment groups. Overall, the odds of improvement in clinical status were higher in the 5-day Remdesivir group at Day 11 when compared to those receiving only SOC (odds ratio, 1.65; [95% CI, 1.09 to 2.48]; p=0.017). The odds of improvement in clinical status with the 10-day treatment group when compared to those receiving only SOC were not statistically significantly different (odds ratio, 1.31; [95% CI 0.88 to 1.95]; p=0.183). At Day 28, mortality was ≤ 2% in all treatment groups.
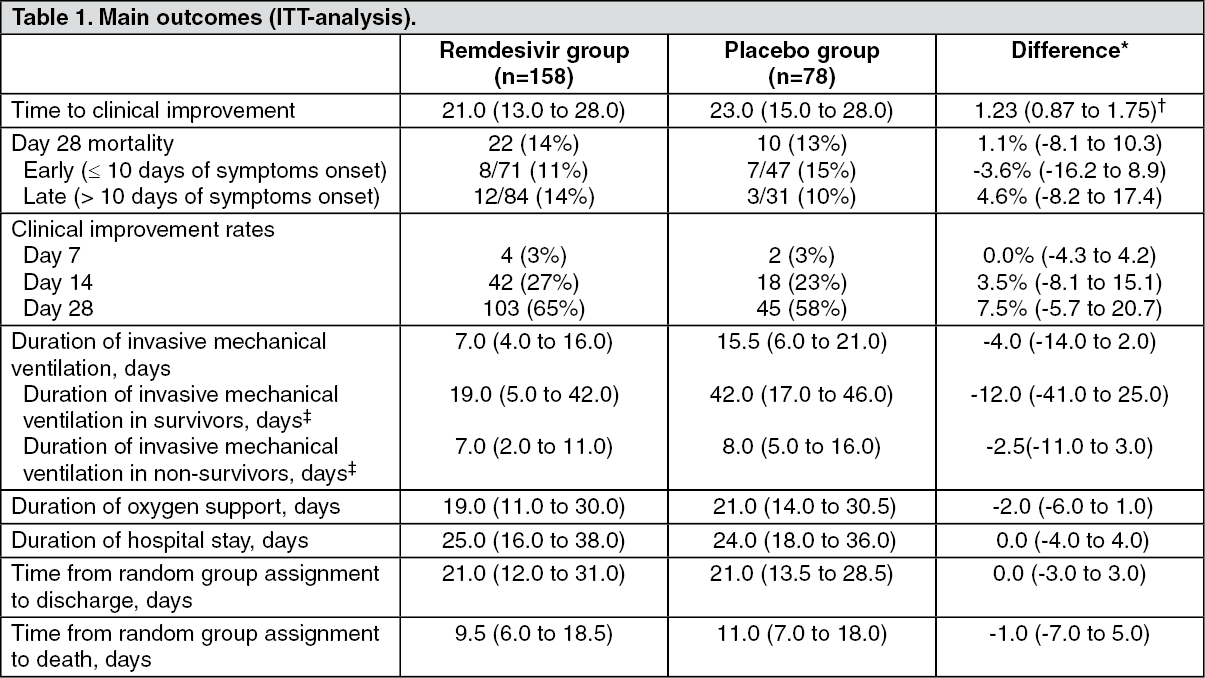
Study GS-US-540-5758 in Hospitalized China Subjects with Severe COVID-19: A randomized, double-blind, placebo control, and multi center clinical trial in China (Study GSUS-540-5758) of hospitalized adult subjects who confirmed SARS-CoV-2 infection with Pneumonia confirmed by chest imaging or had oxygen saturation of 94% or lower on room air, OR, ratio of arterial oxygen partial pressure to fractional inspired oxygen of 300 mmHg or less.
One hundred and fifty-eight subjects received Remdesivir for 10 days (200 mg on the first day and continued with 100 mg Day 10) compared with seventy eight subject received placebo.
The primary endpoint was time to clinical improvement within 28 days after randomization, which defined as a two-point reduction in patients' admission status on a six-point ordinal scale (1, Discharged or having reached discharge criteria (defined as clinical recovery-ie, normalization of pyrexia, respiratory rate <24 breaths per minute, saturation of peripheral oxygen >94% on room air, and relief of cough, all maintained for at least 72 h); 2, Hospital admission but no requiring oxygen therapy; 3, Hospital admission for oxygen therapy (but not requiring high-flow oxygen therapy); 4, Hospital admission for noninvasive ventilation or high-flow oxygen therapy; 5, Hospital admission for extracorporeal membrane oxygenation or mechanical ventilation; and 6, Death), or live discharge from the hospital, whichever came first. The secondary endpoint was 28-day all-cause mortality. The time to clinical improvement in Remdesivir group were not statistically significant different (21 vs 23 days; 95% CI: 0.87 to 1.75). The mortality were also not statistically significant different between Remdesivir and placebo group (14% vs 13%; 95% CI: -8.1 to 10.3). The main outcomes of this trial are presented in the table as follows: (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePharmacokinetics: The pharmacokinetic properties of Remdesivir has been investigated in healthy volunteers.
No pharmacokinetic data is available from patients with COVID-19.
Absorption: The pharmacokinetic properties of Remdesivir and the predominant circulating metabolite GS-441524 have been evaluated in healthy adult subjects. Following intravenous administration of Remdesivir adult dosage regimen, peak plasma concentration was observed at end of infusion, regardless of dose level, and declined rapidly thereafter with a half-life of approximately 1 hour. Peak plasma concentrations of GS-441524 were observed at 1.5 to 2.0 hours post start of a 30 minutes infusion.
Distribution: Remdesivir is approximately 88% bound to human plasma proteins. Protein binding of GS-441524 was low (2% bound) in human plasma. After a single 150 mg dose of [14C]-Remdesivir in healthy subjects, the blood to plasma ratio of 14C-radioactivity was approximately 0.68 at 15 minutes from start of infusion, increased over time reaching ratio of 1.0 at 5 hours, indicating differential distribution of Remdesivir and its metabolites to plasma or cellular components of blood.
Biotransformation: Remdesivir is extensively metabolized to the pharmacologically active nucleoside analog triphosphate GS-443902 (formed intracellularly). The metabolic activation pathway involves hydrolysis by esterases, which leads to the formation of the intermediate metabolite, GS-704277. Phosphoramidate cleavage followed by phosphorylation forms the active triphosphate, GS-443902. Dephosphorylation of all phosphorylated metabolites can result in the formation of nucleoside metabolite GS-441524 that itself is not efficiently re-phosphorylated. The human mass balance study also indicates presence of a currently unidentified major metabolite (M27) in plasma.
Elimination: Following a single 150 mg IV dose of [14C]-Remdesivir, mean total recovery of the dose was 92%, consisting of approximately 74% and 18% recovered in urine and feces, respectively. The majority of the Remdesivir dose recovered in urine was GS-441524 (49%), while 10% was recovered as Remdesivir. These data indicate that renal clearance is the major elimination pathway for GS-441524. The median terminal half-lives of Remdesivir and GS-441524 were approximately 1 and 27 hours, respectively.
Other special populations: Gender, race and age: Pharmacokinetic differences for gender, race, and age have not been evaluated.
Paediatric patients: The pharmacokinetics in paediatric patients have not been evaluated.
Renal impairment: The pharmacokinetics of Remdesivir and GS-441524 in renal impairment has not been evaluated. Remdesivir is not cleared unchanged in urine to any substantial extent, but its main metabolite GS-441524 is renally cleared and the metabolite levels in plasma may theoretically increase in patients with impaired renal function. The excipient betadex sulfobutyl ether sodium is renally cleared and accumulates in patients with decreased renal function. Remdesivir should not be used in patients with eGFR <30 mL/min.
Hepatic impairment: The pharmacokinetics of Remdesivir and GS-441524 in hepatic impairment has not been evaluated. The role of the liver in the metabolism of Remdesivir is unknown.
Preclinical Studies: Toxicology: Following intravenous administration (slow bolus) of Remdesivir to rhesus monkeys and rats, severe renal toxicity occurred after short treatment durations. In male rhesus monkeys at dosage levels of 5, 10, and 20 mg/kg/day for 7 days resulted, at all dose levels, in increased mean urea nitrogen and increased mean creatinine, renal tubular atrophy, and basophilia and casts, and an unscheduled death of one animal at the 20 mg/kg/day dose level. In rats, dosage levels of >3 mg/kg/day for up to 4 weeks resulted in findings indicative of kidney injury and/or dysfunction.
Systemic exposures (AUC) of the predominant circulating metabolite of Remdesivir (GS-441524) were 0.1 times (monkeys at 5 mg/kg/day) and 0.3 times (rat at 3 mg/kg/day) the exposure in humans at the RHD. An unidentified major metabolite (M27) was shown to be present in human plasma. The exposure of M27 in rhesus monkeys and rats is unknown. Animal studies may therefore not be informative of potential risks associated with this metabolite.
Carcinogenesis: Long-term animal studies to evaluate the carcinogenic potential of Remdesivir have not been performed.
Mutagenesis: Remdesivir was not genotoxic in a battery of assays, including bacterial mutagenicity, chromosome aberration using human peripheral blood lymphocytes, and in vivo rat micronucleus assays.
Reproductive toxicity: In female rats, decreases in corpora lutea, numbers of implantation sites, and viable embryos, were seen when Remdesivir was administered intravenously daily at a systemically toxic dose (10 mg/kg/day) 14 days prior to mating and during conception; exposures of the predominant circulating metabolite (GS-441524) were 1.3 times the exposure in humans at the RHD. There were no effects on female reproductive performance (mating, fertility, and conception) at this dose level.
In rats and rabbits, Remdesivir demonstrated no adverse effect on embryofoetal development when administered to pregnant animals at systemic exposures (AUC) of the predominant circulating metabolite of Remdesivir (GS-441524) that were up to 4 times the exposure in humans at the recommended human dose (RHD).
In rats, there were no adverse effects on pre- and post-natal development at systemic exposures (AUC) of the predominant circulating metabolite of Remdesivir (GS-441524) that were similar to the exposure in humans at the recommended human dose (RHD).
It is unknown if the active nucleoside analog triphosphate GS-443902 and the unidentified major human metabolite M27 are formed in rats and rabbits. The reproductive toxicity studies may therefore not be informative of potential risks associated with these metabolites.