


 Sign Out
Sign Out




Durvalumab is a fully human, high affinity, immunoglobulin G1 kappa (IgG1κ) monoclonal antibody that blocks the interaction of PD-L1 with PD-1 and CD80 (B7.1). Durvalumab does not induce antibody dependent cell-mediated cytotoxicity (ADCC). Blockade of PD-L1/PD-1 and PDL1/ CD80 interactions enhances antitumour immune responses. These antitumour responses may result in tumour elimination.
In preclinical studies, PD-L1 blockade by durvalumab led to increased T-cell activation and decreased tumour size in xenograft mouse models of human melanoma and/or pancreatic cancer cells as well as mouse syngeneic colorectal cancer.
Clinical trials: Non-small cell lung cancer (NSCLC): Randomised, placebo-controlled phase 3 study in patients with locally advanced, unresectable NSCLC after chemoradiation (PACIFIC study): The efficacy of IMFINZI was evaluated in the PACIFIC study, a randomised, double-blind, placebo-controlled, multicentre study in 713 patients with histologically or cytologically confirmed locally advanced, unresectable NSCLC. Patients had completed at least 2 cycles of definitive platinum-based chemotherapy with radiation therapy within 1 to 42 days prior to initiation of the study and had an ECOG performance status of 0 or 1. Ninety-two percent of patients had received a total dose of 54 to 66 Gy of radiation. The study excluded patients who had progressed following chemoradiation therapy, patients with prior exposure to any anti-PD-1 or anti-PD-L1 antibody, patients with active or prior documented autoimmune disease within 2 years of initiation of the study; a history of immunodeficiency; a history of severe immune-mediated adverse reactions; medical conditions that required systemic immunosuppression (except physiological dose of systemic corticosteroids); active tuberculosis or hepatitis B or C or HIV infection or patients receiving live attenuated vaccine within 30 days before or after the start of IMFINZI. Patients were randomised 2:1 to receive 10 mg/kg IMFINZI (n=476) or 10 mg/kg placebo (n=237) via intravenous infusion every 2 weeks for up to 12 months or until unacceptable toxicity or confirmed disease progression. Randomisation was stratified by gender, age (<65 years vs. ≥ 65 years) and smoking status (smoker vs. non- smoker). Patients with disease control at 12 months were given the option to be re-treated upon disease progression. Tumour assessments were conducted every 8 weeks for the first 12 months and then every 12 weeks thereafter.
Patients were enrolled regardless of their tumour PD-L1 expression level. Where available, archival tumour tissue specimens taken prior to chemoradiation therapy were retrospectively tested for PD-L1 expression on tumour cells (TC) using the VENTANA PD-L1 (SP263) IHC assay. Of the 713 patients randomised, 63% of patients provided a tissue sample of sufficient quality and quantity to determine PD-L1 expression and 37% were unknown.
The demographics and baseline disease characteristics were well balanced between study arms. Baseline demographics of the overall study population were as follows: male (70%), age ≥ 65 years (45%), white (69%), Asian (27%), other (4%), current smoker (16%), past-smoker (75%), and never smoker (9%), WHO/ECOG PS 0 (49%), WHO/ECOG PS 1 (51%). Disease characteristics were as follows: Stage IIIA (53%), Stage IIIB (45%), histological sub-groups of squamous (46%), non-squamous (54%). Of 451 patients with PD L1 expression available, 67% were TC ≥ 1% [PD-L1 TC 1-24% (32%), PD L1 TC ≥ 25% (35%)] and 33% were TC < 1%.
The two primary endpoints of the study were progression-free survival (PFS) and overall survival (OS) of IMFINZI vs. placebo. Secondary efficacy endpoints included PFS at 12 months (PFS 12) and 18 months (PFS 18) from randomisation and Time from Randomisation to Second Progression (PFS2). PFS was assessed by Blinded Independent Central Review (BICR) according to RECIST 1.1.
The study demonstrated a statistically significant improvement in PFS and OS in the IMFINZI-treated group compared with the placebo group (see Table 1 and Figures 1 and 2).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image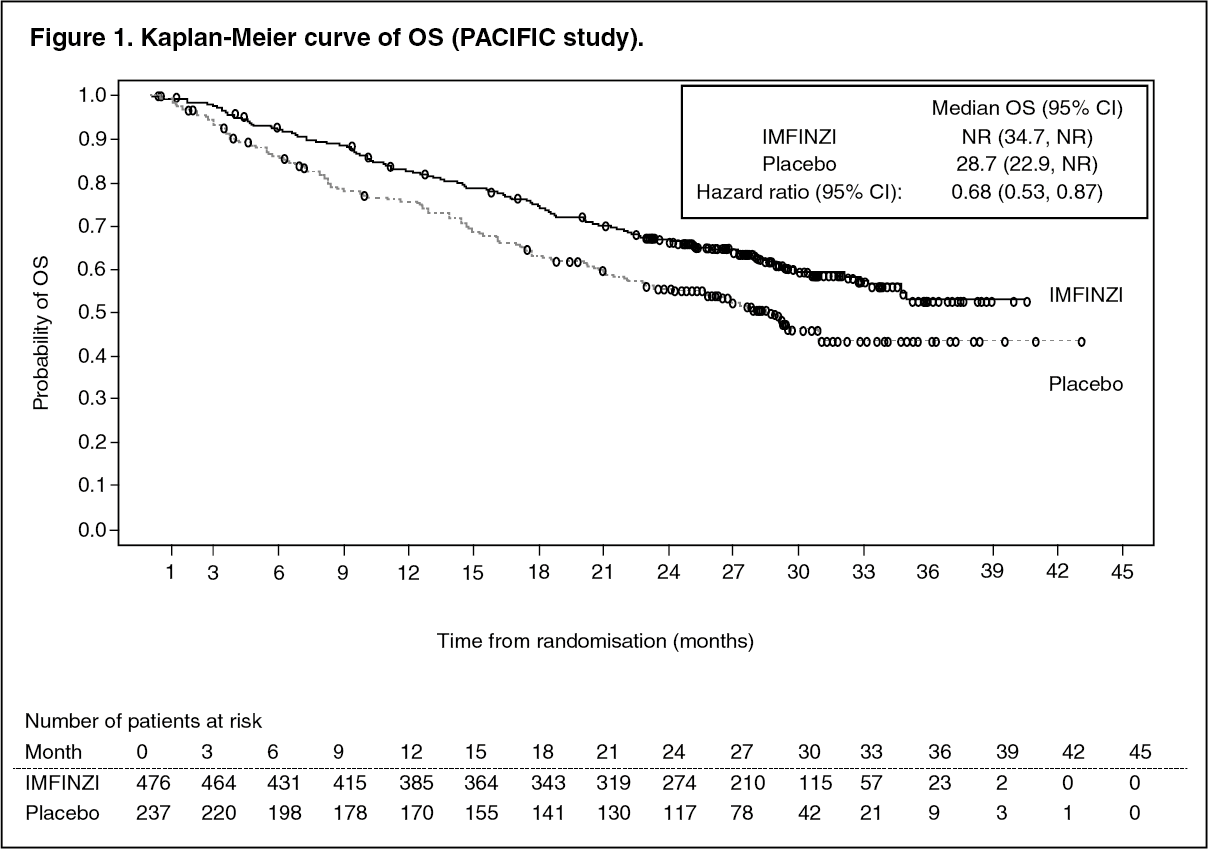 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe improvements in PFS and OS in favour of patients receiving IMFINZI compared to those receiving placebo were consistently observed in all predefined subgroups analysed, including ethnicity, age, gender, smoking history, EGFR mutation status and histology. ALK mutation status was not analysed in this study.
Post-hoc subgroup analysis by PD-L1 expression: Additional subgroup analyses were conducted to evaluate the efficacy by tumour PD-L1 expression (≥ 25%, 1-24%, ≥ 1%, < 1%) and for patients whose PD-L1 status could not be established (PD-L1 unknown). PFS and OS results are summarised in Figures 3 and 4. Overall the safety profile of durvalumab in PD-L1 TC ≥1% subgroup was consistent with the intent to treat population, as was the PD-L1 TC <1% subgroup. (See Figures 3 and 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image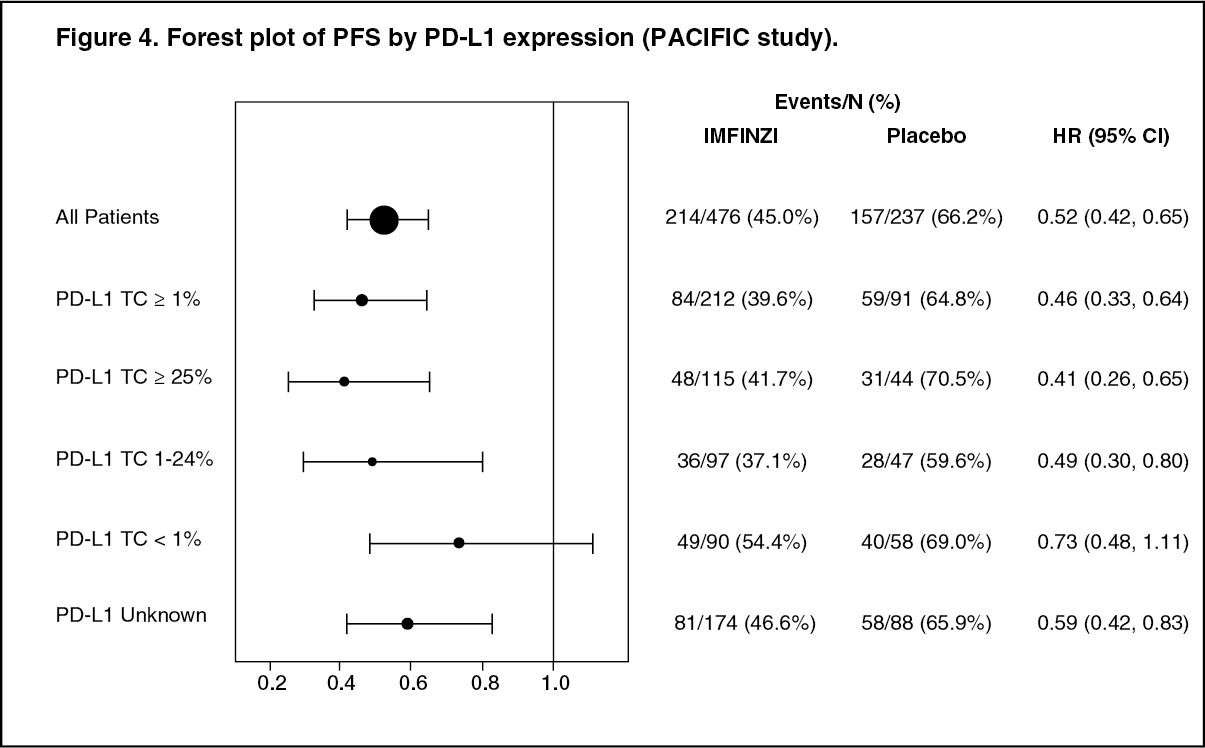 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePatient reported outcomes: Patient-reported symptoms, function and health-related quality of life (HRQoL) were collected using the EORTC QLQ-C30 and its lung cancer module (EORTC QLQ-LC13). The LC13 and C30 were assessed at baseline and every 4 weeks for the first 8 weeks, then every 8 weeks until completion of the treatment period or discontinuation of study drug due to toxicity or disease progression. Compliance was similar between the IMFINZI and placebo treatment groups (83% vs 85.1% overall of evaluable forms completed).
At baseline, no differences in patient reported symptoms, function or HRQoL were observed between IMFINZI and placebo groups. Throughout the duration of the study to week 48, there was no clinically meaningful difference between IMFINZI and placebo groups in symptoms, functioning and HRQoL (as assessed by a difference of greater than or equal to 10 points).
SCLC-CASPIAN Study: CASPIAN was a study designed to evaluate the efficacy of IMFINZI with or without tremelimumab in combination with etoposide and either carboplatin or cisplatin. CASPIAN was a randomized, open-label, multicenter study in 805 treatment naïve ES-SCLC patients with WHO/ECOG Performance status of 0 or 1, suitable to receive a platinum-based chemotherapy regimen as first-line treatment for SCLC, with life expectancy ≥12 weeks, at least one target lesion by RECIST 1.1 and adequate organ and bone marrow function. Patients with asymptomatic or treated brain metastases were eligible. The study excluded patients with a history of chest radiation therapy; a history of active primary immunodeficiency; autoimmune disorders including paraneoplastic syndrome (PNS); active or prior documented autoimmune or inflammatory disorders; use of systemic immunosuppressants within 14 days before the first dose of the treatment except physiological dose of systemic corticosteroids; active tuberculosis or hepatitis B or C or HIV infection; or patients receiving live attenuated vaccine within 30 days before or after the start of IMFINZI.
Randomisation was stratified by the planned platinum-based therapy in cycle 1 (carboplatin or cisplatin).
Patients were randomised 1:1:1 to receive: Arm 1: IMFINZI 1500 mg + tremelimumab 75 mg + etoposide and either carboplatin or cisplatin; Arm 2: IMFINZI 1500 mg + etoposide and either carboplatin or cisplatin; Arm 3: Either carboplatin (AUC 5 or 6 mg/mL/min) or cisplatin (75-80 mg/m2) on Day 1 and etoposide (80-100 mg/m2) intravenously on Days 1, 2, and 3 of each 21-day cycle for between 4-6 cycles.
For patients randomised to Arm 1 and 2, etoposide and either carboplatin or cisplatin was limited to 4 cycles on an every 3 week schedule subsequent to randomisation. IMFINZI monotherapy continued until disease progression or unacceptable toxicity. Administration of IMFINZI monotherapy was permitted beyond disease progression if the patient was clinically stable and deriving clinical benefit as determined by the investigator.
Patients randomised to Arm 3, were permitted to receive a total of up to 6 cycles of etoposide and either carboplatin or cisplatin. After completion of chemotherapy, prophylactic cranial irradiation (PCI) was permitted only in Arm 3 per investigator discretion.
Tumour assessments were conducted at Week 6 and Week 12 from the date of randomisation, and then every 8 weeks until confirmed objective disease progression. Survival assessments were conducted every 2 months following treatment discontinuation.
The primary endpoints of the study were Overall Survival (OS) of IMFINZI + chemotherapy (Arm 2) vs. chemotherapy alone (Arm 3) and IMFINZI + tremelimumab + chemotherapy (Arm 1) vs. chemotherapy alone (Arm 3). The key secondary endpoint was progression-free survival (PFS). Other secondary endpoints were Objective Response Rate (ORR), OS and PFS landmarks and Patient-Reported Outcomes (PRO). PFS and ORR were assessed using Investigator assessments according to RECIST v1.1.
The demographics and baseline disease characteristics were well balanced between the two study arms (268 patients in Arm 2 and 269 patients in Arm 3). Baseline demographics of the overall study population were as follows: male (69.6%), age ≥ 65 years (39.6%), median age 63 years (range: 28 to 82 years), white (83.8%), Asian (14.5%), black or African American (0.9%), other (0.6 %), non-Hispanic or Latino (96.1%), current or past-smoker (93.1%), never smoker (6.9%), WHO/ECOG PS 0 (35.2%), WHO/ECOG PS 1 (64.8%), Stage IV 90.3%, 24.6% of the patients received cisplatin and 74.1% of the patients received carboplatin. In Arm 3, 56.8% of the patients received 6 cycles of etoposide + platinum and 7.8% of the patients received PCI.
At a planned interim (primary) analysis the study demonstrated a statistically significant improvement in OS with IMFINZI + etoposide + platinum (Arm 2) vs. etoposide + platinum alone (Arm 3) [HR=0.73 (95% CI: 0.591, 0.909), p=0.0047]. IMFINZI + etoposide + platinum demonstrated an improvement in PFS vs. etoposide + platinum alone [HR=0.78 (95% CI: 0.645, 0.936). See Table 2 and Figures 5 and 6.
In the planned follow-up analysis (median: 25.1 months), IMFINZI + etoposide + platinum (Arm 2) vs. etoposide + platinum (Arm 3) continued to demonstrate improved OS. The OS, PFS, ORR and DoR results from the planned follow-up analysis are summarized in Table 5; Kaplan-Meier curves for OS and PFS are presented in Figures 5 and 6. (See Table 2 and Figures 5 and 6.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSubgroup analysis: The improvements in OS in favor of patients receiving IMFINZI + chemotherapy compared to those receiving chemotherapy alone, were consistently observed across the prespecified subgroups based on demographics, geographical region, carboplatin or cisplatin use and disease characteristics.
Pharmacokinetics: The pharmacokinetics (PK) of durvalumab was assessed for both IMFINZI monotherapy and in combination with chemotherapy.
The pharmacokinetics of IMFINZI was studied in 2903 patients with solid tumours with doses ranging from 0.1 to 20 mg/kg administered once every two, three or four weeks as monotherapy.
Distribution: PK exposure increased more than dose-proportionally (non-linear PK) at doses <3 mg/kg and dose proportionally (linear PK) at doses ≥ 3 mg/kg. Steady state was achieved at approximately 16 weeks. Based on population PK analysis that included 1878 patients who received durvalumab monotherapy in the dose range of ≥ 10 mg/kg Q2W, the steady state volume of distribution (Vss) was 5.64 L.
Excretion: Durvalumab clearance (CL) decreased over time resulting in a geometric mean steady state clearance (CLss) of 8.16 mL/h at Day 365; the decrease in CLss was not considered clinically relevant. The terminal half-life (t1/2), based on baseline CL, was approximately 18 days. There was no clinically meaningful difference between the PK of durvalumab as a single agent and in combination with chemotherapy.
Special Populations: Age (19-96 years), body weight (34-149 kg), gender, positive anti-drug antibody (ADA) status, albumin levels, LDH levels, creatinine levels, soluble PD-L1, tumour type, race, mild renal impairment (creatinine clearance (CRCL) 60 to 89 mL/min), moderate renal impairment (creatinine clearance (CRCL) 30 to 59 mL/min), mild hepatic impairment (bilirubin ≤ ULN and AST > ULN or bilirubin >1.0 to 1.5 × ULN and any AST) and ECOG/WHO status had no clinically significant effect on the pharmacokinetics of durvalumab.
The effect of severe renal impairment (CRCL 15 to 29 mL/min) or moderate (bilirubin >1.5 to 3 x ULN and any AST) or severe (bilirubin >3.0 x ULN and any AST) hepatic impairment on the pharmacokinetics of durvalumab is unknown; however, as IgG monoclonal antibodies are not primarily cleared via hepatic pathways, a change in hepatic function is not expected to influence durvalumab exposure.
Laboratory abnormalities: In patients treated with durvalumab monotherapy, the proportion of patients who experienced a shift from baseline to a Grade 3 or 4 laboratory abnormality was as follows: 2.4% for alanine aminotransferase increased, 3.6% for aspartate aminotransferase increased, 0.5% for blood creatinine increased, 5.7% for amylase increased and 5.6% for lipase increased. The proportion of patients who experienced a TSH shift from baseline that was ≤ ULN to any grade > ULN was 18.8% and a TSH shift from baseline that was ≥ LLN to any grade < LLN was 18.1%.
In patients treated with durvalumab in combination with chemotherapy, the proportion of patients who experienced a shift from baseline to a Grade 3 or 4 laboratory abnormality was as follows: 4.9% for alanine aminotransferase increased, 4.6% for aspartate aminotransferase increased, 3.4% for blood creatinine increased, 4.8% for amylase increased and 8.1% for lipase increased. The proportion of patients who experienced a TSH shift from baseline that was ≤ ULN to any grade > ULN was 17.7% and a TSH shift from baseline that was ≥ LLN to any grade < LLN was 31.3%.
Immunogenicity: As with all therapeutic proteins, there is a potential for immunogenicity. Of the 2280 patients who were treated with IMFINZI 10 mg/kg every 2 weeks and evaluable for the presence of anti-drug antibodies (ADA). Sixty nine patients (3.0%) tested positive for treatment emergent ADA. Neutralising antibodies against durvalumab were detected in 0.5% (12/2280) patients. The presence of ADAs did not have a clinically relevant effect on pharmacokinetics or safety.
In the CASPIAN study, of the 201 patients who were treated with IMFINZI 1500 mg every 3 weeks in combination with chemotherapy and evaluable for the presence of ADAs, 0 (0%) patients tested positive for treatment-emergent ADAs. The impact of treatment-emergent ADA on pharmacokinetics and clinical safety of durvalumab was not evaluable as no patient samples tested positive for treatment-emergent durvalumab ADA.
Immunogenicity assay results are highly dependent on several factors, including assay sensitivity and specificity, assay methodology, sample handling, timing of sample collection, concomitant medications and underlying disease.
For these reasons, comparison of incidence of antibodies to IMFINZI with the incidence of antibodies to other products may be misleading.
Toxicology: Preclinical safety data: Genotoxicity: The genotoxic potential of durvalumab has not been evaluated. As a large protein molecule, durvalumab is not expected to interact directly with DNA or other chromosomal material.
Carcinogenicity: The carcinogenic potential of durvalumab has not been evaluated.