


 Sign Out
Sign Out



Pharmacology: Pharmacodynamics: Cardiac Electrophysiology: GIOTRIF at doses of 50 mg daily did not result in significant prolongation of the QTcF interval after single and multiple administrations in patients with relapsed or refractory solid tumours. There were no cardiac safety findings of clinical concern suggesting that GIOTRIF does not have a relevant effect on the QTcF interval.
Mechanism of Action: Afatinib is a potent and selective, irreversible ErbB Family Blocker. Afatinib covalently binds to and irreversibly blocks signalling from all homo- and heterodimers formed by the ErbB family members EGFR (ErbB1), HER2 (ErbB2), ErbB3 and ErbB4.
Pharmacodynamic Effects: Aberrant ErbB signalling triggered by, for instance, EGFR mutations and/or amplification, HER2 amplification or mutation and/or ErbB ligand overexpression contributes to the malignant phenotype in subsets of patients across multiple cancer types. In preclinical disease models with ErbB pathway deregulation, afatinib as a single agent effectively blocks ErbB receptor signalling resulting in tumour growth inhibition or tumour regression. NSCLC models with either L858R or Del 19 EGFR mutations are particularly sensitive to afatinib treatment. Afatinib retains significant anti-tumour activity in NSCLC cell lines in vitro and tumour models in vivo (xenografts or transgenic models) driven by mutant EGFR isoforms known to be resistant to the reversible EGFR inhibitors erlotinib and gefitinib such as T790M.
Clinical Trials: GIOTRIF in Patients Naïve to EGFR TKI Treatment: LUX-Lung 3 (1200.32): In the first-line setting, the efficacy and safety of GIOTRIF in patients with EGFR mutation-positive locally advanced or metastatic NSCLC (stage IIIB or IV) were assessed in a global, randomised, multicentre, open-label trial (LUX-Lung 3). Patients naive to prior systemic treatment for their advanced or metastatic disease were screened for the presence of 29 different EGFR mutations using a polymerase chain reaction (PCR) based method (TheraScreen: EGFR29 Mutation Kit, Qiagen Manchester Ltd). Patients (N=345) were randomised (2:1) to receive GIOTRIF 40 mg orally once daily (N=230) or up to 6 cycles pemetrexed/cisplatin (N=115). Randomisation was stratified according to EGFR mutation status (L858R; Del 19; other) and race (Asian; non-Asian). Mean duration of treatment was 336 and 105 days for the GIOTRIF and chemotherapy arms, respectively.
The primary endpoint of PFS (independent review, 221 events) showed statistically significant improvement in the median PFS between patients treated with GIOTRIF and patients treated with chemotherapy (11.1 vs. 6.9 months). When comparing the pre-specified subgroup of common (L858R or Del 19) EGFR mutations, the difference in PFS was further pronounced (13.6 vs. 6.9 months). The percentage of patients alive and without progression (PFS rate) at 12 months was 46.5% in patients treated with GIOTRIF and 22% in patients treated with chemotherapy for the overall trial population, and 51.1% vs. 21.4% in the subgroup of common mutations.
The subgroup of "other" (uncommon) mutations was small (N=37; 11%) and genetically heterogeneous (10 different molecular subtypes with unequal distribution between the treatment groups) thereby limiting the value and interpretation of the pooled statistical analyses in this subset. Individual responses and prolonged disease stabilisation were observed in some patients with uncommon mutations.
The Kaplan-Meier curve of primary PFS analysis is shown in figure and efficacy results are summarised in Table 1. At the same time of primary analysis a total of 45 (20%) patients treated with GIOTRIF and 3 (3%) patients treated with chemotherapy were known to be alive and progression-free and are censored in figure. (See figure and Table 1).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image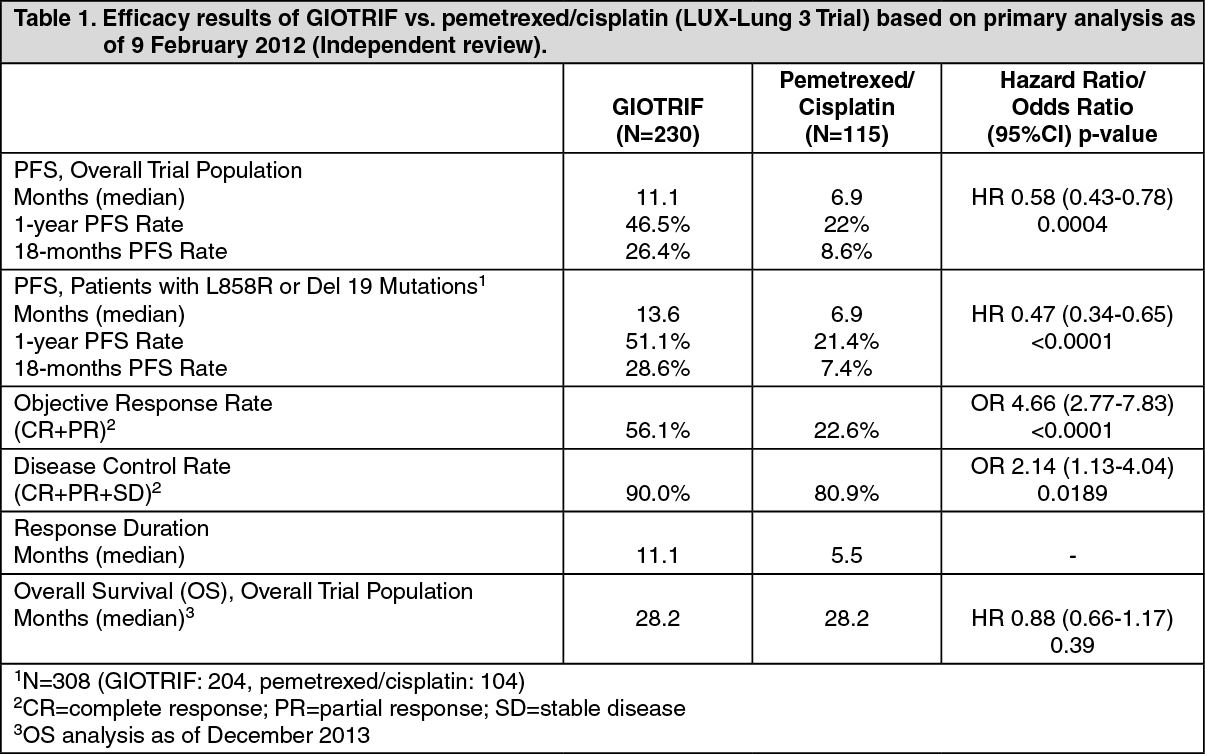 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePFS benefit was accompanied by improvement in disease-related symptoms, as measured by the European Organization for Research and Treatment of Cancer (EORTC) Quality of Life Questionnaires (QLQ-C30 and QLQ-LC13). GIOTRIF significantly delayed the time to deterioration for pre-specified symptoms of cough (HR 0.6; p=0.0072) and dyspnoea (HR 0.68; p=0.0145) by more than 7 months when compared with chemotherapy. Time to deterioration of pain was also longer with GIOTRIF but did not reach statistical significance (HR 0.83; p=0.1913). Significantly more patients treated with GIOTRIF compared with those treated with chemotherapy had improvement for dyspnoea (64% vs. 50%; p=0.0103). A trend favouring GIOTRIF was observed for pain (59% vs. 48%; p=0.0513), with individual items of pain reaching significance ('Have pain': 56.0% vs. 40.0%; p=0.0095; 'Pain in chest': 51.0% vs. 37.0%; p=0.0184; 'Pain in arm or shoulder': 41.0% vs. 26.0%; p=0.0103). For cough, numerically more patients improved on GIOTRIF (67% vs. 60%; p=0.2444).
Mean scores over time for health-related quality of life (HRQoL) were measured using the EORTC QLQC30. Mean scores over time for overall quality of life and global health status were significantly better for GIOTRIF compared with chemotherapy. Mean scores were significantly better in 3 of the 5 functioning domains (physical, role, cognitive) and showed no difference in the emotional and social functioning domains.
LUX-Lung 2 (1200.22): Lux-Lung 2 was an open label single arm Phase II trial which investigated the efficacy & safety of GIOTRIF in 129 EGFR TKI-naïve patients with locally advanced or metastatic lung adenocarcinoma (stage IIIB or IV) with EGFR mutations. Patients were enrolled in the first-line (N=61) or second-line setting (N=68) (i.e. after failure of 1 prior chemotherapy regimen). Patients were centrally screened for EGFR mutations.
The primary endpoint was ORR. Secondary endpoints included PFS, DCR and OS.
In 61 patients treated in the first-line setting, confirmed ORR was 65.6% and DCR was 86.9% according to independent review. The median PFS was 12.0 months by independent review and 15.6 months by investigator assessment. Median OS was not reached in the first-line population. Efficacy was similarly high in the group of patients who had received prior chemotherapy (N=68; ORR 57.4%; PFS by independent review 8 months and by investigator assessment 10.5 months; DCR 77.9%). Median OS in the second line patients was 23.3 months (95% CI 18.5-38).
Pharmacokinetics: Absorption and Distribution: Following oral administration of GIOTRIF, maximum concentrations (Cmax) of afatinib are observed approximately 2 to 5 hours post dose. Mean Cmax and AUC0-∞ values increased slightly more than 50 mg GIOTRIF. Systemic exposure to afatinib is decreased by 50% (Cmax) and 39% (AUC0-∞), when administered with a high-fat meal compared with administration in the fasted state. Based on population pharmacokinetic data derived from clinical trials in various tumour types, an average decrease of 26% in AUCΤ,ss was observed when food was consumed within 3 hours before or 1 hour after taking GIOTRIF. Therefore, food should not be consumed for at least 3 hours before and at least 1 hour after taking GIOTRIF (see Dosage & Administration and Interactions). After administration of GIOTRIF, the mean relative bioavailability was 92% (adjusted gMean ratio of AUC0-∞) when compared to an oral solution.
In vitro binding of afatinib to human plasma proteins is approximately 95%.
The volume of distribution was 1,940 L for single dose treatment and 2,770 L at steady state. The absolute bioavailability of GIOTRIF is unknown.
Metabolism and Excretion: Enzyme-catalyzed metabolic reactions play a negligible role for afatinib in vivo. Covalent adducts to proteins are the major circulating metabolites of afatinib. Following administration of an oral solution of 15 mg afatinib, 85.4% of the dose was recovered in the faeces and 4.3% in urine. The parent compound afatinib accounted for 88% of the recovered dose. The apparent terminal half-life is 37 hours. Steady state plasma concentrations of afatinib are achieved within 8 days of multiple dosing of afatinib resulting in an accumulation of 2.77-fold (AUC) and 2.11-fold (Cmax).
Renal Impairment: Less than 5% of a single dose of afatinib is excreted via the kidneys. The safety, pharmacokinetics and efficacy of GIOTRIF have not been studied specifically in patients with renal impairment. Based on population pharmacokinetic data derived from clinical trials in various tumour types, no dose adjustments appear necessary in patients with mild or moderate renal impairment (see Population Pharmacokinetic Analysis in Special Populations as follows and Dosage & Administration).
Hepatic Impairment: Afatinib is eliminated mainly by biliary/faecal excretion. Subjects with mild (Child-Pugh A) or moderate (Child-Pugh B) hepatic impairment had similar exposure in comparison to healthy volunteers following a single dose of 50 mg GIOTRIF. This is consistent with population pharmacokinetic data derived from clinical trials in various tumour types (see Population Pharmacokinetic Analysis in Special Populations as follows). No starting dose adjustments appear necessary in patients with mild or moderate hepatic impairment (see Dosage & Administration). The pharmacokinetics of afatinib had not been studied in subjects with severe (Child-Pugh C) hepatic dysfunction (see Precautions).
Population Pharmacokinetic Analysis in Special Populations: A population pharmacokinetic analysis was performed in 927 cancer patients (764 with NSCLC) receiving GIOTRIF monotherapy. No starting dose adjustment is considered necessary for any of the following covariates tested.
Age: No significant impact of age (range: 28-87 years) the pharmacokinetics of afatinib could be observed.
Body Weight: Plasma exposure (AUCt,ss) was increased by 26% for a 42 kg patient (2.5th percentile) and decreased by 22% for a 95 kg patient (97.5th percentile) relative to a patietn weighing 62 kg (median body weight of patients in the overall patient population).
Gender: Female patients had a 15% higher plasma exposure (AUCΤ,ss, body weight corrected) than male patients.
Race: There was no statistically significant difference in afatinib pharmacokinetics between Asian and Caucasian patients. Also, no obvious difference in pharmacokinetics for American Indian/Alaska native or Black patients could be detected based on the limited data available in these populations (6 and 9 out of 927 patients included in the analysis, respectively).
Renal Impairment: Exposure to GIOTRIF moderately increased with lowering the creatinine clearance (CrCl), i.e. for a patient with a CrCl of 60 or 30 mL/min exposure (AUCΤ,ss) to afatinib increased by 13% and 42%, respectively, and decreased by 6% and 20% for a patient with CrCl of 90 or 120 mL, respectively, compared to a patient with the CrCl of 79 mL/min (median CrCl of patients in the overall patient population analysed).
Hepatic Impairment: Patients with mild and moderate hepatic impairment as identified by abnormal liver tests did not correlate with any significant change in afatinib exposure.
Other Patient Characteristics/Intrinsic Factors: Other patient characteristics/intrinsic factors found with a significant impact on afatinib exposure were: ECOG performance score, lactate dehydrogenase levels, alkaline phosphatase levels and total protein. The individual effect sizes of these covariates were considered not clinically relevant. Smoking history, alcohol consumption, or presence of liver metastases had no significant impact on the pharmacokinetics of afatinib.
Pharmacokinetic Drug Interactions: Drug Transporters: P-glycoprotein (P-gp): Effect of P-gp Inhibitors and Inducers on Afatinib: Two trials were conducted to assess the effect of ritonavir, a potent inhibitor of P-gp, on the pharmacokinetics of afatinib. In one trial, the relative bioavailability of afatinib was investigated when ritonavir (200 mg b.i.d for 3 days) was given either simultaneously or 6 hours after a single dose of 40 mg GIOTRIF. The relative bioavailability of afatinib was 119% (AUC0-∞) and 104% (Cmax) when administered simultaneously with ritonavir and 111% (AUC0-∞) and 105% (Cmax) when ritonavir was administered 6 hours after GIOTRIF. In a second trial when ritonavir (200 mg b.i.d for 3 days) was administered 1 hour before a single dose of 20 mg GIOTRIF, exposure to afatinib increased by 48% (AUC0-∞) and 39% (Cmax) (see Dosage & Administration, Precautions, and Interactions).
Pre-treatment with rifampicin (600 mg q.d. for 7 days), a potent inducer of P-gp, decreased the plasma exposure to afatinib by 34% (AUC0-∞) and 22% (Cmax) after administration of a single dose of 40 mg GIOTRIF (see Precautions and Interactions).
Effect of Afatinib on P-gp Substrates: Based on in vitro data, afatinib is a moderate inhibitor of P-gp. It is considered unlikely that GIOTRIF treatment will result in changes of the plasma concentrations of other P-gp substrates.
Breast Cancer Resistance Protein (BCRP): In vitro studies indicated that afatinib is a substrate and an inhibitor of the transporter BCRP.
Drug Uptake Transport Systems: In vitro data indicated that drug-drug interactions with afatinib due to inhibition of OATB1B1, OATP1B3, OATP2B1, OAT1, OAT3, OCT1, OCT2, and OCT3 transporters are considered unlikely.
Drug Metabolising Enzymes: Cytochrome P450 (CYP) Enzymes: Effect of CYP Enzymes Inducers and Inhibitors on Afatinib: In vitro data indicated that drug-drug interactions with afatinib due to inhibition or induction of CYP enzymes by concomitant medicines are considered unlikely. In humans, it was found that enzyme-catalyzed metabolic reactions play a negligible role for the metabolism of afatinib. Approximately 2% of the afatinib dose was metabolized by FMO3 and the CYP3A4-dependent N-demethylation was too low to be quantitatively detected.
Effect of Afatinib on CYP Enzymes: Afatinib is not an inhibitor or an inducer of CYP enzymes. Therefore, GIOTRIF is unlikely to affect the metabolism of other medicines that are dependent on CYP enzymes.
UDP-glucuronosyltransferase 1A1 (UGT1A1): In vitro data indicated that drug-drug interactions with afatinib due to inhibition of UGT1A1 are considered unlikely.
Toxicology: Oral administration of single doses to mice and rats indicated a low acute toxic potential of afatinib. In oral repeated-dose studies for up to 26 weeks in rats or 52 weeks in minipigs, the main effects were identified in the skin (dermal changes, epithelial atrophy and folliculitis in rats), the gastrointestinal tract (diarrhoea, erosions in the stomach, epithelial atrophy in rats and minipigs) and the kidneys (papillary necrosis in rats). Depending on the finding, these changes occurred at exposures below, in the range of or above clinically relevant levels. Additionally, in various organs pharmacodynamically mediated atrophy of epithelia was observed in both species.
Reproduction Toxicity: Based on the mechanism of action, GIOTRIF has the potential to cause foetal harm. The embryofoetal development studies performed on afatinib revealed no indication of teratogenicity up to dose levels including maternal death. Changes identified were restricted to skeletal alterations consisting of incomplete ossifications/unossified elements (rat) and abortions at maternally toxic dose, reduced foetal weights as well as mainly visceral and dermal variations (rabbit). The respective total systemic exposure (AUC) was either slightly above (2.2 times in rats) or below (0.3 times in rabbits) compared with levels in patients.
Radiolabelled afatinib administered orally to pregnant rats on Day 11 of lactation was excreted into milk of the dams. The average concentrations in milk at time points 1 h and 6 h post dose were approximately 80- and 150-fold above the respective concentration in plasma.
A fertility study in male and female rats by the oral route up to the maximum tolerated dose revealed no significant impact on fertility. The total systemic exposure (AUC0-24) that could be achieved in male and female rats was in the range or less than that observed in patients (1.3 times and 0.51 times, respectively).
A study in rats by the oral route up to the maximum tolerated doses revealed no significant impact on pre-/postnatal development. Effects were limited to lower birth weight and body weight gain of offspring but without materially affecting the attainment of developmental landmarks, sexual maturation or performance with behavioural assessments. The highest total systemic exposure (AUC0-24) that could be achieved in female rats was less than that observed in patients (0.23 times).
Phototoxicity: An in vitro 3T3 phototoxicity test with afatinib was performed. It was concluded that GIOTRIF may have phototoxicity potential.
Carcinogenicity: Carcinogenicity studies have not been conducted with GIOTRIF.
A marginal response to afatinib was observed in a single tester strain of a bacterial (Ames) mutagenicity assay. However, no mutagenic or genotoxic potential could be identified in an in vitro chromosomal aberration test at non-cytotoxic concentrations as well as the in vivo bone marrow micronucleus assay, the in vivo Comet assay and an in vivo 4-week oral mutation study in the Muta Mouse.