Pharmacology: Mechanism of Action: Ticagrelor and its major metabolite reversibly interact with the platelet P2Y12 ADP-receptor to prevent signal transduction and platelet activation. Ticagrelor and its active metabolite are approximately equipotent.
Pharmacodynamics: The inhibition of platelet aggregation (IPA) by ticagrelor and clopidogrel was compared in a 6 week study examining both acute and chronic platelet inhibition effects in response to 20 μM ADP as the platelet aggregation agonist.
The onset of IPA was evaluated on Day 1 of the study following loading doses of ticagrelor 180 mg or clopidogrel 600 mg. As shown in Figure 1, IPA was higher in the ticagrelor group at all time points. The maximum IPA effect of ticagrelor was reached at around 2 hours and was maintained for at least 8 hours.
The offset of IPA was examined after 6 weeks on ticagrelor 90 mg twice daily or clopidogrel 75 mg daily, again in response to 20 μM ADP.
As shown in Figure 2, mean maximum IPA following the last dose of ticagrelor was 88% and 62% for clopidogrel. The insert in figure 2 shows that after 24 hours, IPA in the ticagrelor group (58%) was similar to IPA in clopidogrel group (52%), indicating that patients who miss a dose of ticagrelor would still maintain IPA similar to the trough IPA of patients treated with clopidogrel. After 5 days, IPA in the ticagrelor group was similar to IPA in the placebo group. It is not known how either bleeding risk or thrombotic risk track with IPA, for either ticagrelor or clopidogrel. (See Figures 1 and 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Transitioning from clopidogrel to BRILINTA resulted in an absolute IPA increase of 26.4% and from BRILINTA to clopidogrel resulted in an absolute IPA decrease of 24.5%. Patients can be transitioned from clopidogrel to BRILINTA without interruption of antiplatelet effect (see Dosage & Administration).
Adenosine mechanism (ENT-1): Ticagrelor increased plasma adenosine concentrations in ACS patients and has been shown to augment several physiological responses to adenosine. Adenosine is a vasodilator; ticagrelor has been shown to augment adenosine-induced coronary blood flow increases in healthy volunteers and ACS patients. Adenosine is an endogenous platelet inhibitor; ticagrelor has been shown to augment adenosine-mediated inhibition of platelet aggregation in addition to platelet inhibition due to its P2Y12 antagonism. Adenosine has been linked to the cardioprotective effect of preconditioning; ticagrelor has been shown to reduce infarct size via an adenosine-mediated mechanism in a rat model of reperfusion injury. Adenosine also induces dyspnoea; ticagrelor has been shown to augment adenosine-induced dyspnoea in healthy volunteers. Thus, the dyspnoea observed in some patients taking ticagrelor (see Adverse Reactions) may partly or completely be mediated by adenosine.
Pharmacokinetics: Ticagrelor demonstrates dose proportional pharmacokinetics, which are similar in patients and healthy volunteers.
Absorption: Absorption of ticagrelor occurs with a median tmax of 1.5 h (range 1.0-4.0). The formation of the major circulating metabolite AR-C124910XX (active) from ticagrelor occurs with a median tmax of 2.5 h (range 1.5-5.0).
The mean absolute bioavailability of ticagrelor is about 36%, (range 30%-42%). Ingestion of a high-fat meal had no effect on ticagrelor Cmax, but resulted in a 21% increase in AUC. The Cmax of its major metabolite was decreased by 22% with no change in AUC. BRILINTA can be taken with or without food.
Brilinta as crushed tablets mixed in water, given orally or administered through a nasogastric tube into the stomach, is bioequivalent to whole tablets (AUC and Cmax within 80-125% for ticagrelor and the active metabolite), with a median tmax of 1.0 hour (range 1.0-4.0) for ticagrelor and 2.0 hours (range 1.0-8.0) for AR-C124910XX.
Distribution: The steady state volume of distribution of ticagrelor is 88 L. Ticagrelor and the active metabolite are extensively bound to human plasma proteins (>99%).
Metabolism: CYP3A4 is the major enzyme responsible for ticagrelor metabolism and the formation of its major active metabolite. Ticagrelor and its major active metabolite are weak P-glycoprotein (P-gp) substrates and inhibitors. The systemic exposure to the active metabolite is approximately 30-40% of the exposure of ticagrelor.
Excretion: The primary route of ticagrelor elimination is hepatic metabolism. When radiolabeled ticagrelor is administered, the mean recovery of radioactivity is approximately 84% (58% in feces, 26% in urine). Recoveries of ticagrelor and the active metabolite in urine were both less than 1% of the dose. The primary route of elimination for the major metabolite of ticagrelor is most likely to be biliary secretion. The mean t1/2 is approximately 7 hours for ticagrelor and 9 hours for the active metabolite.
Special Populations: The effects of age, gender, ethnicity, renal impairment and mild hepatic impairment on the pharmacokinetics of ticagrelor are presented in Figure 3. Effects are modest and do not require dose adjustment. (See Figure 3.)
Click on icon to see table/diagram/image
Pediatric: Ticagrelor has not been evaluated in a pediatric population (see Precautions).
Body Weight: No dose adjustment is necessary for ticagrelor based on weight.
Smoking: Habitual smoking increased population mean clearance of ticagrelor by approximately 22% when compared to non-smokers. No dose adjustment is necessary for ticagrelor based on smoking status.
Renal impairment: In patients with end stage renal disease on haemodialysis AUC and Cmax of ticagrelor 90 mg administered on a day without dialysis were 38% and 51% higher compared to subjects with normal renal function. A similar increase in exposure was observed when ticagrelor was administered immediately prior to dialysis (49% and 61%, respectively) showing that ticagrelor is not dialysable. Exposure of the active metabolite increased to a lesser extent (AUC 13-14% and Cmax 17-36%). The inhibition of platelet aggregation (IPA) effect of ticagrelor was independent of dialysis in patients with end stage renal disease and similar to subjects with normal renal function
Effects of Other Drugs on BRILINTA : CYP3A4 is the major enzyme responsible for ticagrelor metabolism and the formation of its major active metabolite. The effects of other drugs on the pharmacokinetics of ticagrelor are presented in Figure 4 as change relative to ticagrelor given alone (test/reference). Strong CYP3A inhibitors (e.g., ketoconazole, itraconazole, and clarithromycin) substantially increase ticagrelor exposure. Moderate CYP3A inhibitors have lesser effects (e.g., diltiazem). CYP3A inducers (e.g., rifampin) substantially reduce ticagrelor blood levels. P-gp inhibitors (e.g. cyclosporine) increase ticagrelor exposure. (See Figure 4.)
Click on icon to see table/diagram/image
Effects of BRILINTA on Other Drugs: In vitro metabolism studies demonstrate that ticagrelor and its major active metabolite are weak inhibitors of CYP3A4, potential activators of CYP3A5 and inhibitors of the P-gp transporter. Ticagrelor and AR-C124910XX were shown to have no inhibitory effect on human CYP1A2, CYP2C19, and CYP2E1 activity. For specific
in vivo effects on the pharmacokinetics of simvastatin, atorvastatin, ethinyl estradiol, levonorgesterol, tolbutamide, digoxin and cyclosporine, see Figure 5. (See Figure 5.)
Click on icon to see table/diagram/image
Pharmacogenetics: In a genetic substudy cohort of PLATO, the rate of thrombotic CV events in the BRILINTA arm did not depend on CYP2C19 loss of function status.
Toxicology: Nonclinical Toxicology: Carcinogenesis: Ticagrelor was not carcinogenic in the mouse at doses up to 250 mg/kg/day or in the male rat at doses up to 120 mg/kg/day (19 and 15 times MRHD of 90 mg twice daily on the basis of AUC, respectively). Uterine carcinomas, uterine adenocarcinomas and hepatocellular adenomas were seen in female rats at doses of 180 mg/kg/day (29-fold the maximally recommended dose of 90 mg twice daily on the basis of AUC), whereas 60 mg/kg/day (8-fold the MRHD based on AUC) was not carcinogenic in female rats.
Mutagenesis: Ticagrelor did not demonstrate genotoxicity when tested in the Ames bacterial mutagenicity test, mouse lymphoma assay and the rat micronucleus test. The active O-demethylated metabolite did not demonstrate genotoxicity in the Ames assay and mouse lymphoma assay.
Impairment of Fertility: Ticagrelor had no effect on male fertility at doses up to 180 mg/kg/day or on female fertility at doses up to 200 mg/kg/day (>15-fold the MRHD on the basis of AUC). Doses of ≥10 mg/kg/day given to female rats caused an increased incidence of irregular duration estrus cycles (1.5-fold the MRHD based on AUC).
Clinical Studies: Acute Coronary Syndromes and Secondary Prevention after Myocardial Infarction: PLATO: PLATO was a randomized double-blind study comparing BRILINTA (N=9333) to clopidogrel (N=9291), both given in combination with aspirin and other standard therapy, in patients with acute coronary syndromes (ACS), who presented within 24 hours of onset of the most recent episode of chest pain or symptoms. The study's primary endpoint was the composite of first occurrence of cardiovascular death, non-fatal MI (excluding silent MI), or non-fatal stroke.
Patients who had already been treated with clopidogrel could be enrolled and randomized to either study treatment. Patients with previous intracranial hemorrhage, gastrointestinal bleeding within the past 6 months, or with known bleeding diathesis or coagulation disorder were excluded. Patients taking anticoagulants were excluded from participating and patients who developed an indication for anticoagulation during the trial were discontinued from study drug. Patients could be included whether there was intent to manage the ACS medically or invasively, but patient randomization was not stratified by this intent.
All patients randomized to BRILINTA received a loading dose of 180 mg followed by a maintenance dose of 90 mg twice daily. Patients in the clopidogrel arm were treated with an initial loading dose of clopidogrel 300 mg, if clopidogrel therapy had not already been given. Patients undergoing PCI could receive an additional 300 mg of clopidogrel at investigator discretion. A daily maintenance dose of aspirin 75-100 mg was recommended, but higher maintenance doses of aspirin were allowed according to local judgment. Patients were treated for at least 6 months and for up to 12 months.
PLATO patients were predominantly male (72%) and Caucasian (92%). About 43% of patients were >65 years and 15% were >75 years. Median exposure to study drug was 277 days. About half of the patients received pre-study clopidogrel and about 99% of the patients received aspirin at some time during PLATO. About 35% of patients were receiving a statin at baseline and 93% received a statin sometime during PLATO.
Table 1 shows the study results for the primary composite endpoint and the contribution of each component to the primary endpoint. Separate secondary endpoint analyses are shown for the overall occurrence of CV death, MI, and stroke and overall mortality. (See Table 1 and Figure 6.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The curves separate by 30 days (RRR 12%) and continue to diverge throughout the 12 month treatment period (RRR 16%). Among 11289 patients with PCI receiving any stent during PLATO, there was a lower risk of stent thrombosis (1.3% for adjudicated "definite") than with clopidogrel (1.9%) (HR 0.67, 95% CI 0.50-0.91; p=0.009). The results were similar for drug-eluting and bare metal stents.
A wide range of demographic, concurrent baseline medications, and other treatment differences were examined for their influence on outcome. Many of these are shown in Figure 6. Such analyses must be interpreted cautiously, as differences can reflect the play of chance among a large number of analyses. Most of the analyses show effects consistent with the overall results, but there are two marked exceptions: A finding of heterogeneity by region and a strong influence of the maintenance dose of aspirin. These are considered further as follows.
Most of the characteristics shown are baseline characteristics, but some reflect post-randomization determinations (eg, final diagnosis, aspirin maintenance dose, use of PCI). (See Figure 7.)
Click on icon to see table/diagram/image
Regional Differences:
Results in the rest of the world compared to effects in North America (US and Canada) show a smaller effect in North America, numerically inferior to the control and driven by the US subset. The statistical test for the US/non-US comparison is statistically significant (p=0.009), and the same trend is present for both CV death and non-fatal MI. The individual results and nominal p-values, like all subset analyses, need cautious interpretation, and they could represent chance findings. The consistency of the differences in both the CV mortality and non-fatal MI components, however, supports the possibility that the finding is reliable.
A wide variety of baseline and procedural differences between the US and non-US (including intended invasive vs. planned medical management, use of GPIIb/IIIa inhibitors, use of drug eluting vs bare-metal stents) were examined to see if they could account for regional differences, but with one exception, aspirin maintenance dose, these differences did not appear to lead to differences in outcome.
Aspirin Dose: The PLATO protocol left the choice of aspirin maintenance dose up to the investigator and use patterns were different in US sites from sites outside of the US. About 8% of non-US investigators administered aspirin doses above 100 mg, and about 2% administered doses above 300 mg. In the US, 57% of patients received doses above 100 mg and 54% received doses above 300 mg. Overall results favored BRILINTA when used with low maintenance doses (≤100 mg) of aspirin, and results analyzed by aspirin dose were similar in the US and elsewhere. Figure 8 shows overall results by median aspirin dose. Figure 8 shows results by region and dose. (See Figure 8.)
Click on icon to see table/diagram/image
Like any unplanned subset analysis, especially one where the characteristic is not a true baseline characteristic (but may be determined by usual investigator practice), the above analyses must be treated with caution. It is notable, however, that aspirin dose predicts outcome in both regions with a similar pattern, and that the pattern is similar for the two major components of the primary endpoint, CV death and non-fatal MI.
Despite the need to treat such results cautiously, there appears to be good reason to restrict aspirin maintenance dosage accompanying ticagrelor to 100 mg. Higher doses do not have an established benefit in the ACS setting, and there is a strong suggestion that use of such doses reduces the effectiveness of BRILINTA.
PEGASUS: The PEGASUS TIMI-54 study was a 21162-patient, randomized, double-blind, placebo-controlled, parallel-group study. Two doses of ticagrelor, either 90 mg twice daily or 60 mg twice daily, co-administered with 75-150 mg of aspirin, were compared to aspirin therapy alone in patients with history of MI. The primary endpoint was the composite of first occurrence of CV death, non-fatal MI and non-fatal stroke. CV death and all-cause mortality were assessed as secondary endpoints.
Patients were eligible to participate if they were ≥50 years old, with a history of MI 1 to 3 years prior to randomization, and had at least one of the following risk factors for thrombotic cardiovascular events: age ≥65 years, diabetes mellitus requiring medication, at least one other prior MI, evidence of multivessel coronary artery disease, or creatinine clearance <60 mL/min. Patients could be randomized regardless of their prior ADP receptor blocker therapy or a lapse in therapy. Patients requiring or who were expected to require renal dialysis during the study were excluded. Patients with any previous intracranial hemorrhage, gastrointestinal bleeding within the past 6 months, or with known bleeding diathesis or coagulation disorder were excluded. Patients taking anticoagulants were excluded from participating and patients who developed an indication for anticoagulation during the trial were discontinued from study drug. A small number of patients with a history of stroke were included. Based on information external to PEGASUS, 102 patients with a history of stroke (90 of whom received study drug) were terminated early and no further such patients were enrolled.
Patients were treated for at least 12 months and up to 48 months with a median follow up time of 33 months.
Patients were predominantly male (76%) Caucasian (87%) with a mean age of 65 years, and 99.8% of patients received prior Aspirin therapy. See Table 2 for key baseline features. (See Table 2.)
Click on icon to see table/diagram/image
The Kaplan-Meier curve (Figure 9) shows time to first occurrence of the primary composite endpoint of CV death, non-fatal MI or non-fatal stroke. (See Figure 9.)
Click on icon to see table/diagram/image
Both the 60 mg and 90 mg regimens of BRILINTA in combination with aspirin were superior to aspirin alone in reducing the incidence of CV death, MI or stroke. The absolute risk reductions for BRILINTA plus aspirin vs. aspirin alone were 1.27% and 1.19% for the 60 and 90 mg regimens, respectively. Although the efficacy profiles of the two regimens were similar, the lower dose had lower risks of bleeding and dyspnea.
Table 3 shows the results for the 60 mg plus aspirin regimen vs. aspirin alone. (See Table 3.)
Click on icon to see table/diagram/image
In PEGASUS, the RRR for the composite endpoint from 1 to 360 days (17% RRR) and from 361 days and onwards (16% RRR) was similar. The treatment effect of BRILINTA 60 mg twice daily over aspirin was consistent across pre-defined subgroups, see Figure 10. (See Figure 10).
Click on icon to see table/diagram/image





 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image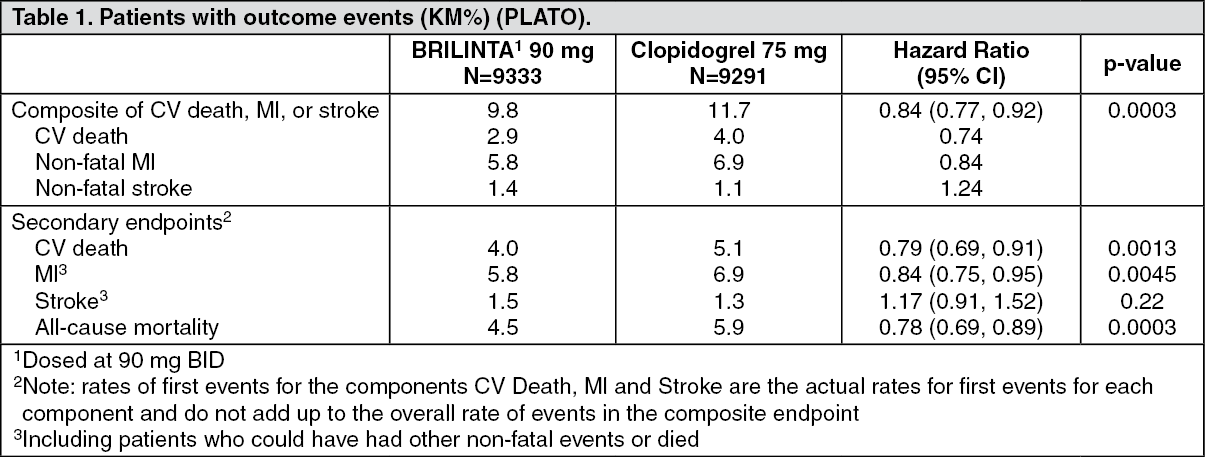 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
