


 Sign Out
Sign Out



Pharmacodynamics: Amphetamines block the reuptake of norepinephrine and dopamine into the presynaptic neuron and increase the release of these monoamines into the extraneuronal space. The parent drug, lisdexamfetamine, does not bind to the sites responsible for the reuptake of norepinephrine and dopamine in vitro.
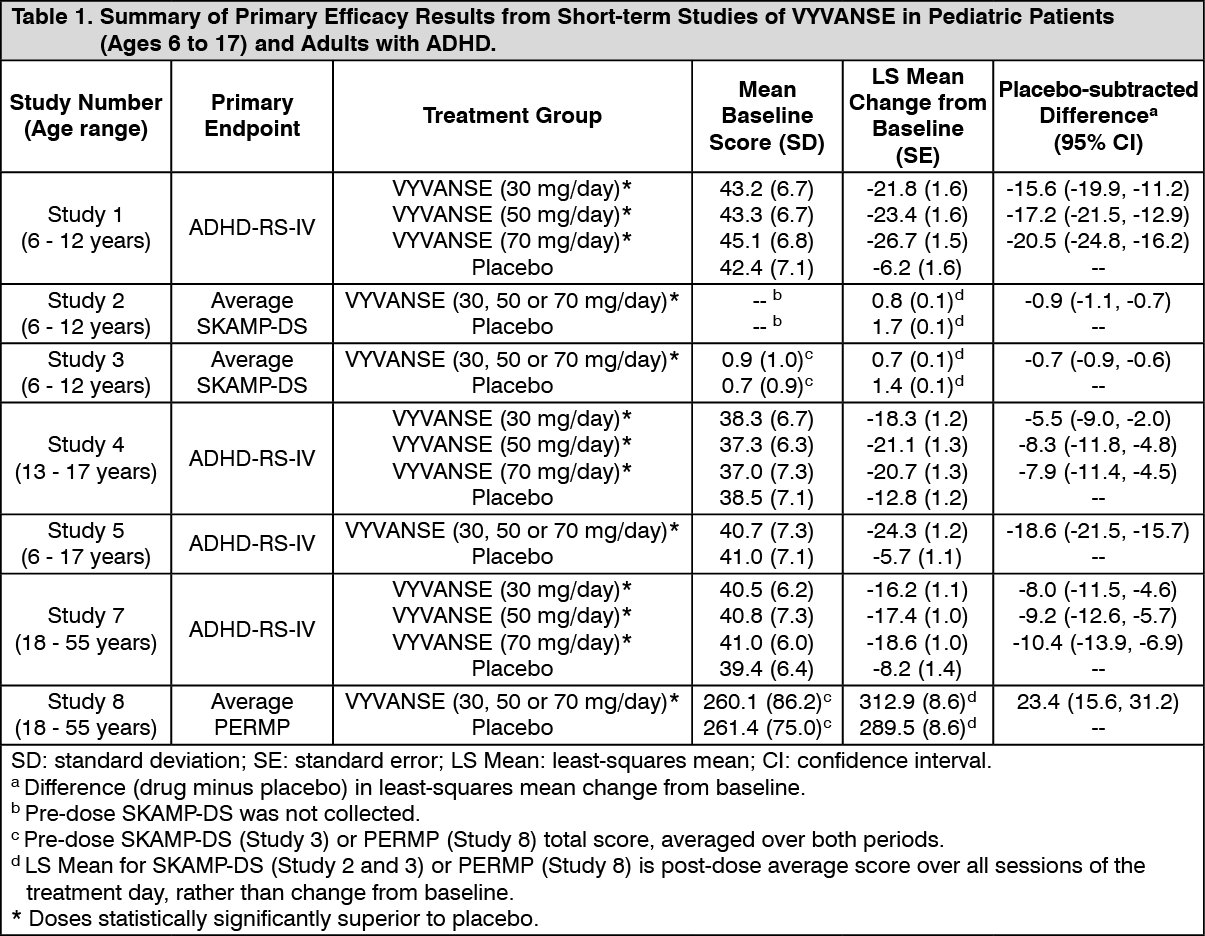
Clinical Studies: Attention Deficit Hyperactivity Disorder (ADHD): Pediatric Patients Ages 6 to 12 Years with ADHD: A double-blind, randomized, placebo-controlled, parallel-group study (Study 1) was conducted in pediatric patients ages 6 to 12 years (N=290) who met DSM-IV criteria for ADHD (either the combined type or the hyperactive-impulsive type). Patients were randomized to receive final doses of 30 mg, 50 mg, or 70 mg of VYVANSE or placebo once daily in the morning for a total of four weeks of treatment. All patients receiving VYVANSE were initiated on 30 mg for the first week of treatment. Patients assigned to the 50 mg and 70 mg dose groups were titrated by 20 mg per week until they achieved their assigned dose. The primary efficacy outcome was change in Total Score from baseline to endpoint in investigator ratings on the ADHD Rating Scale (ADHD-RS), an 18-item questionnaire with a score range of 0-54 points that measures the core symptoms of ADHD which includes both hyperactive/impulsive and inattentive subscales. Endpoint was defined as the last post-randomization treatment week (i.e., Weeks 1 through 4) for which a valid score was obtained. All VYVANSE dose groups were superior to placebo in the primary efficacy outcome. Mean effects at all doses were similar; however, the highest dose (70 mg/day) was numerically superior to both lower doses (Study 1 in Table 1). The effects were maintained throughout the day based on parent ratings (Conners' Parent Rating Scale) in the morning (approximately 10 am), afternoon (approximately 2 pm), and early evening (approximately 6 pm). (See Table 1.)
A double-blind, placebo-controlled, randomized, cross-over design, analog classroom study (Study 2) was conducted in pediatric patients ages 6 to 12 years (N=52) who met DSM-IV criteria for ADHD (either the combined type or the hyperactive-impulsive type). Following a 3-week open-label dose optimization with Adderall XR, patients were randomly assigned to continue their optimized dose of Adderall XR (10 mg, 20 mg, or 30 mg), VYVANSE (30 mg, 50 mg, or 70 mg), or placebo once daily in the morning for 1 week each treatment. Efficacy assessments were conducted at 1, 2, 3, 4.5, 6, 8, 10, and 12 hours post-dose using the Swanson, Kotkin, Agler, M.Flynn, and Pelham Deportment scores (SKAMP-DS), a 4-item subscale of the SKAMP with scores ranging from 0 to 24 points that measures deportment problems leading to classroom disruptions. A significant difference in patient behavior, based upon the average of investigator ratings on the SKAMP-DS across the 8 assessments, were observed between patients when they received VYVANSE compared to patients when they received placebo (Study 2 in Table 1). The drug effect reached statistical significance from hours 2 to 12 post-dose, but was not significant at 1 hour. (See Table 1.)
A second double-blind, placebo-controlled, randomized, cross-over design, analog classroom study (Study 3) was conducted in pediatric patients ages 6 to 12 years (N=129) who met DSM-IV criteria for ADHD (either the combined type or the hyperactive-impulsive type). Following a 4-week open-label dose optimization with VYVANSE (30 mg, 50 mg, 70 mg), patients were randomly assigned to continue their optimized dose of VYVANSE or placebo once daily in the morning for 1 week each treatment. A significant difference in patient behavior, based upon the average of investigator ratings on the SKAMP-Deportment scores across all 7 assessments conducted at 1.5, 2.5, 5.0, 7.5, 10.0, 12.0, and 13.0 hours post-dose, were observed between patients when they received VYVANSE compared to patients when they received placebo (Study 3 in Table 1, Figure 1). (See Table 1 and Figure 1.)
Pediatric Patients Ages 13 to 17 Years with ADHD: A double-blind, randomized, placebo-controlled, parallel-group study (Study 4) was conducted in pediatric patients ages 13 to 17 years (N=314) who met DSM-IV criteria for ADHD. In this study, patients were randomized in a 1:1:1:1 ratio to a daily morning dose of VYVANSE (30 mg/day, 50 mg/day or 70 mg/day) or placebo for a total of four weeks of treatment. All patients receiving VYVANSE were initiated on 30 mg for the first week of treatment. Patients assigned to the 50 mg and 70 mg dose groups were titrated by 20 mg per week until they achieved their assigned dose. The primary efficacy outcome was change in Total Score from baseline to endpoint in investigator ratings on the ADHD Rating Scale (ADHD-RS). Endpoint was defined as the last post-randomization treatment week (i.e., Weeks 1 through 4) for which a valid score was obtained. All VYVANSE dose groups were superior to placebo in the primary efficacy outcome (Study 4 in Table 1). (See Table 1.)
Pediatric Patients Ages 6 to 17 Years: Short-Term Treatment in ADHD: A double-blind, randomized, placebo- and active-controlled parallel-group, dose-optimization study (Study 5) was conducted in pediatric patients ages 6 to 17 years (N=336) who met DSM-IV criteria for ADHD. In this eight-week study, patients were randomized to a daily morning dose of VYVANSE (30, 50 or 70 mg/day), an active control, or placebo (1:1:1). The study consisted of a Screening and Washout Period (up to 42 days), a 7-week Double-blind Evaluation Period (consisting of a 4-week Dose-Optimization Period followed by a 3-week Dose-Maintenance Period), and a 1-week Washout and Follow-up Period. During the Dose-Optimization Period, subjects were titrated until an optimal dose, based on tolerability and investigator's judgment, was reached. VYVANSE showed significantly greater efficacy than placebo. The placebo-adjusted mean reduction from baseline in the ADHD-RS-IV total score was 18.6. Subjects on VYVANSE also showed greater improvement on the Clinical Global Impression-Improvement (CGI-I) rating scale compared to subjects on placebo (Study 5 in Table 1). (See Table 1.)
Pediatric Patients Ages 6 to 17 Years: Maintenance Treatment in ADHD: Maintenance of Efficacy Study (Study 6) - A double-blind, placebo-controlled, randomized withdrawal study was conducted in pediatric patients ages 6 to 17 years (N=276) who met the diagnosis of ADHD (DSM-IV criteria). A total of 276 patients were enrolled into the study, 236 patients participated in Study 5 and 40 subjects directly enrolled. Subjects were treated with open-label VYVANSE for at least 26 weeks prior to being assessed for entry into the randomized withdrawal period. Eligible patients had to demonstrate treatment response as defined by CGI-S <3 and Total Score on the ADHD-RS ≤22. Patients that maintained treatment response for 2 weeks at the end of the open-label treatment period were eligible to be randomized to ongoing treatment with the same dose of VYVANSE (N=78) or switched to placebo (N=79) during the double-blind phase. Patients were observed for relapse (treatment failure) during the 6-week double-blind phase. A significantly lower proportion of treatment failures occurred among VYVANSE subjects (15.8%) compared to placebo (67.5%) at endpoint of the randomized withdrawal period. The endpoint measurement was defined as the last post-randomization treatment week at which a valid ADHD-RS Total Score and CGI-S were observed. Treatment failure was defined as a ≥50% increase (worsening) in the ADHD-RS Total Score and a ≥2-point increase in the CGI-S score compared to scores at entry into the double-blind randomized withdrawal phase. Subjects who withdrew from the randomized withdrawal period and who did not provide efficacy data at their last on-treatment visit were classified as treatment failures (Study 6, Figure 2). (See Figure 2.)
Adults: Short-Term Treatment in ADHD: A double-blind, randomized, placebo-controlled, parallel-group study (Study 7) was conducted in adults ages 18 to 55 (N=420) who met DSM-IV criteria for ADHD. In this study, patients were randomized to receive final doses of 30 mg, 50 mg, or 70 mg of VYVANSE or placebo for a total of four weeks of treatment. All patients receiving VYVANSE were initiated on 30 mg for the first week of treatment. Patients assigned to the 50 mg and 70 mg dose groups were titrated by 20 mg per week until they achieved their assigned dose. The primary efficacy outcome was change in Total Score from baseline to endpoint in investigator ratings on the ADHD Rating Scale (ADHD-RS). Endpoint was defined as the last post-randomization treatment week (i.e., Weeks 1 through 4) for which a valid score was obtained. All VYVANSE dose groups were superior to placebo in the primary efficacy outcome (Study 7 in Table 1). (See Table 1.)
The second study was a multi-center, randomized, double-blind, placebo-controlled, cross-over, modified analog classroom study (Study 8) of VYVANSE to simulate a workplace environment in 142 adults ages 18 to 55 who met DSM-IV-TR criteria for ADHD. There was a 4-week open-label dose-optimization phase with VYVANSE (30 mg/day, 50 mg/day, or 70 mg/day in the morning). Patients were then randomized to one of two treatment sequences: 1) VYVANSE (optimized dose) followed by placebo, each for one week, or 2) placebo followed by VYVANSE, each for one week. Efficacy assessments occurred at the end of each week, using the Permanent Product Measure of Performance (PERMP), a skill-adjusted math test that measures attention in ADHD. PERMP total score results from the sum of the number of math problems attempted plus the number of math problems answered correctly. VYVANSE treatment, compared to placebo, resulted in a statistically significant improvement in attention across all post-dose time points, as measured by average PERMP total scores over the course of one assessment day, as well as at each time point measured. The PERMP assessments were administered at pre-dose (-0.5 hours) and at 2, 4, 8, 10, 12, and 14 hours post-dose (Study 8 in Table 1, Figure 3). (See Table 1 and Figure 3.)
Adults: Maintenance Treatment in ADHD: A double-blind, placebo-controlled, randomized withdrawal design study (Study 9) was conducted in adults ages 18 to 55 (N=123) who had a documented diagnosis of ADHD or met DSM-IV criteria for ADHD. At study entry, patients must have had documentation of treatment with VYVANSE for a minimum of 6 months and had to demonstrate treatment response as defined by Clinical Global Impression Severity (CGI-S) ≤3 and Total Score on the ADHD-RS <22. ADHD-RS Total Score is a measure of core symptoms of ADHD. The CGI-S score assesses the clinician's impression of the patient's current illness state and ranges from 1 (not at all ill) to 7 (extremely ill). Patients that maintained treatment response at Week 3 of the open-label treatment phase (N=116) were eligible to be randomized to ongoing treatment with the same dose of VYVANSE (N=56) or switched to placebo (N=60) during the double-blind phase. Patients were observed for relapse (treatment failure) during the 6-week double-blind phase. The efficacy endpoint was the proportion of patients with treatment failure during the double-blind phase. Treatment failure was defined as a ≥50% increase (worsening) in the ADHD-RS Total Score and ≥2-point increase in the CGI-S score compared to scores at entry into the double-blind phase. Maintenance of efficacy for patients treated with VYVANSE was demonstrated by the significantly lower proportion of patients with treatment failure (9%) compared to patients receiving placebo (75%) at endpoint during the double-blind phase (Study 9, Figure 4). (See Figure 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePharmacokinetics: Pharmacokinetic studies after oral administration of lisdexamfetamine dimesylate have been conducted in healthy adult and pediatric (6 to 12 years) patients with ADHD. After single-dose administration of lisdexamfetamine dimesylate, pharmacokinetics of dextroamphetamine was found to be linear between 30 mg and 70 mg in a pediatric study (6 to 12 years) and between 50 mg and 250 mg in an adult study. Dextroamphetamine pharmacokinetic parameters following administration of lisdexamfetamine dimesylate in adults exhibited low inter-subject (<25%) and intra-subject (<8%) variability. There is no accumulation of lisdexamfetamine and dextroamphetamine at steady state in healthy adults.
Absorption: Following single-dose oral administration of VYVANSE capsule (30 mg, 50 mg, or 70 mg) in patients ages 6 to 12 years with ADHD under fasted conditions, Tmax of lisdexamfetamine and dextroamphetamine was reached at approximately 1 hour and 3.5 hours post-dose, respectively.
Weight/Dose-normalized AUC and Cmax values were the same in pediatric patients ages 6 to 12 years as the adults following single doses of 30 mg to 70 mg VYVANSE capsule.
Effect of food on capsule formulation: Neither food (a high-fat meal or yogurt) nor orange juice affects the observed AUC and Cmax of dextroamphetamine in healthy adults after single-dose oral administration of 70 mg of VYVANSE capsules. Food prolongs Tmax by approximately 1 hour (from 3.8 hours at fasted state to 4.7 hours after a high-fat meal or to 4.2 hours with yogurt). After an 8-hour fast, the AUC for dextroamphetamine following oral administration of lisdexamfetamine dimesylate in solution and as intact capsules were equivalent.
Elimination: Plasma concentrations of unconverted lisdexamfetamine are low and transient, generally becoming non-quantifiable by 8 hours after administration. The plasma elimination half-life of lisdexamfetamine typically averaged less than one hour in volunteers ages 6 years and older. The plasma elimination half-life of dextroamphetamine was approximately 8.6 to 9.5 hours in pediatric patients 6 to 12 years and 10 to 11.3 hours in healthy adults.
Metabolism: Lisdexamfetamine is converted to dextroamphetamine and l-lysine primarily in blood due to the hydrolytic activity of red blood cells after oral administration of lisdexamfetamine dimesylate. In vitro data demonstrated that red blood cells have a high capacity for metabolism of lisdexamfetamine; substantial hydrolysis occurred even at low hematocrit levels (33% of normal). Lisdexamfetamine is not metabolized by cytochrome P450 enzymes.
Excretion: Following oral administration of a 70 mg dose of radiolabeled lisdexamfetamine dimesylate to 6 healthy subjects, approximately 96% of the oral dose radioactivity was recovered in the urine and only 0.3% recovered in the feces over a period of 120 hours. Of the radioactivity recovered in the urine, 42% of the dose was related to amphetamine, 25% to hippuric acid, and 2% to intact lisdexamfetamine.
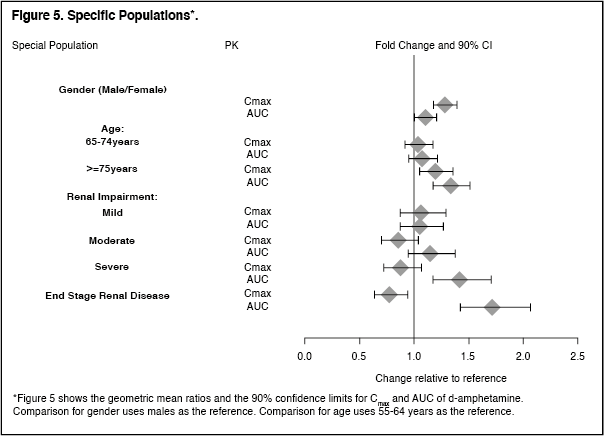
Specific Populations: Exposures of dextroamphetamine in specific populations are summarized in Figure 5. (See Figure 5.)
 Click on icon to see table/diagram/image
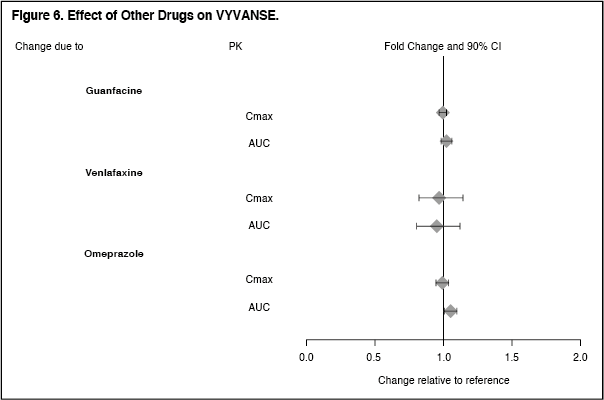
Click on icon to see table/diagram/imageDrug Interaction Studies: Effects of other drugs on the exposures of dextroamphetamine are summarized in Figure 6. (See Figure 6.)
 Click on icon to see table/diagram/image
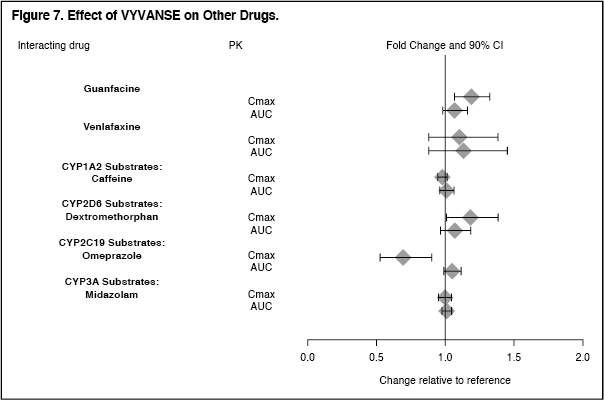
Click on icon to see table/diagram/imageThe effects of VYVANSE on the exposures of other drugs are summarized in Figure 7. (See Figure 7.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageToxicology: Nonclinical Toxicology: Carcinogenesis, Mutagenesis, and Impairment of Fertility: Carcinogenesis: Carcinogenicity studies of lisdexamfetamine dimesylate have not been performed. No evidence of carcinogenicity was found in studies in which d-, l-amphetamine (enantiomer ratio of 1:1) was administered to mice and rats in the diet for 2 years at doses of up to 30 mg/kg/day in male mice, 19 mg/kg/day in female mice, and 5 mg/kg/day in male and female rats.
Mutagenesis: Lisdexamfetamine dimesylate was not clastogenic in the mouse bone marrow micronucleus test in vivo and was negative when tested in the E. coli and S. typhimurium components of the Ames test and in the L5178Y/TK+- mouse lymphoma assay in vitro.
Impairment of Fertility: Amphetamine (d- to l-enantiomer ratio of 3:1) did not adversely affect fertility or early embryonic development in the rat at doses of up to 20 mg/kg/day.
Animal Toxicology and/or Pharmacology: Acute administration of high doses of amphetamine (d- or d,l-) has been shown to produce long-lasting neurotoxic effects, including irreversible nerve fiber damage, in rodents. The significance of these findings to humans is unknown.