Each tablet contains 5 mg or 10 mg of ambrisentan.
Excipient(s) with known effect: 5 mg: Each tablet contains approximately 95 mg of lactose (as monohydrate), approximately 0.25 mg of lecithin (soya) (E322) and approximately 0.11 mg of Allura red AC Aluminium Lake (E129).
10 mg: Each tablet contains approximately 90 mg of lactose (as monohydrate), approximately 0.25 mg of lecithin (soya) (E322) and approximately 0.45 mg of Allura red AC Aluminium Lake (E129).
Excipients/Inactive Ingredients: Tablet core: Lactose monohydrate, Microcrystalline cellulose, Croscarmellose sodium, Magnesium stearate.
Film coat: Polyvinyl alcohol (partially hydrolysed), Talc (E553b), Titanium dioxide (E171), Macrogol / PEG 3350, Lecithin (soya) (E322), Allura red AC Aluminium Lake (E129).
Pharmacotherapeutic group: Anti-hypertensives, other anti-hypertensives. ATC code: C02KX02.
Pharmacology: Pharmacodynamics: Mechanism of action: Ambrisentan is an orally active, propanoic acid-class, ERA selective for the endothelin A (ETA) receptor. Endothelin plays a significant role in the pathophysiology of PAH.
Ambrisentan is a potent (Ki 0.016 nM) and highly selective ETA antagonist (approximately 4000-fold more selective for ETA as compared to ETB).
Ambrisentan blocks the ETA receptor subtype, localized predominantly on vascular smooth muscle cells and cardiac myocytes. This prevents endothelin-mediated activation of second messenger systems that result in vasoconstriction and smooth muscle cell proliferation.
The selectivity of ambrisentan for the ETA over the ETB receptor is expected to retain ETB receptor mediated production of the vasodilators nitric oxide and prostacyclin.
Clinical efficacy and safety: Two randomised, double-blind, multi-centre, placebo controlled, Phase 3 pivotal studies were conducted (ARIES-1 and 2). ARIES-1 included 201 patients and compared ambrisentan 5 mg and 10 mg with placebo. ARIES-2 included 192 patients and compared ambrisentan 2.5 mg and 5 mg with placebo. In both studies, ambrisentan was added to patients' supportive/background medication, which could have included a combination of digoxin, anticoagulants, diuretics, oxygen and vasodilators (calcium channel blockers, ACE inhibitors). Patients enrolled had IPAH or PAH associated with connective tissue disease (PAH-CTD). The majority of patients had WHO functional Class II (38.4%) or Class III (55.0%) symptoms. Patients with pre-existent hepatic disease (cirrhosis or clinically significantly elevated aminotransferases) and patients using other targeted therapy for PAH (e.g. prostanoids) were excluded. Haemodynamic parameters were not assessed in these studies.
The primary endpoint defined for the Phase 3 studies was improvement in exercise capacity assessed by change from baseline in 6 minute walk distance (6MWD) at 12 weeks. In both studies, treatment with ambrisentan resulted in a significant improvement in 6MWD for each dose of ambrisentan.
The placebo-adjusted improvement in mean 6MWD at week 12 compared to baseline was 30.6 m (95% CI: 2.9 to 58.3; p=0.008) and 59.4 m (95% CI: 29.6 to 89.3; p<0.001) for the 5 mg group, in ARIES 1 and 2 respectively. The placebo-adjusted improvement in mean 6MWD at week 12 in patients in the 10 mg group in ARIES-1 was 51.4 m (95% CI: 26.6 to 76.2; p <0.001).
A pre-specified combined analysis of the Phase 3 studies (ARIES-C) was conducted. The placebo-adjusted mean improvement in 6MWD was 44.6 m (95% CI: 24.3 to 64.9; p<0.001) for the 5 mg dose, and 52.5 m (95% CI: 28.8 to 76.2; p<0.001) for the 10 mg dose.
In ARIES-2, ambrisentan (combined dose group) significantly delayed the time to clinical worsening of PAH compared to placebo (p<0.001), the hazard ratio demonstrated an 80% reduction (95% CI: 47% to 92%). The measure included: death, lung transplantation, hospitalisation for PAH, atrial septostomy, addition of other PAH therapeutic agents and early escape criteria. A statistically significant increase (3.41 ± 6.96) was observed for the combined dose group in the physical functioning scale of the SF-36 Health Survey compared with placebo (-0.20 ± 8.14, p=0.005). Treatment with ambrisentan led to a statistically significant improvement in Borg Dyspnea Index (BDI) at week 12 (placebo-adjusted BDI of -1.1 (95% CI: -1.8 to -0.4; p=0.019; combined dose group)).
Long term data: Patients enrolled into ARIES 1 and 2 were eligible to enter a long term open label extension study ARIES E (n=383). The combined mean exposure was approximately 145 ± 80 weeks, and the maximum exposure was approximately 295 weeks. The main primary endpoints of this study were the incidence and severity of adverse events associated with long-term exposure to ambrisentan, including serum LFTs. The safety findings observed with long-term ambrisentan exposure in this study were generally consistent with those observed in the 12 week placebo-controlled studies.
The observed probability of survival for subjects receiving ambrisentan (combined ambrisentan dose group) at 1, 2 and 3 years was 93%, 85% and 79% respectively.
In an open label study (AMB222), ambrisentan was studied in 36 patients to evaluate the incidence of increased serum aminotransferase concentrations in patients who had previously discontinued other ERA therapy due to aminotransferase abnormalities. During a mean of 53 weeks of treatment with ambrisentan, none of the patients enrolled had a confirmed serum ALT>3xULN that required permanent discontinuation of treatment. Fifty percent of patients had increased from 5 mg to 10 mg ambrisentan during this time.
The cumulative incidence of serum aminotransferase abnormalities >3xULN in all Phase 2 and 3 studies (including respective open label extensions) was 17 of 483 subjects over a mean exposure duration of 79.5 weeks. This is an event rate of 2.3 events per 100 patient years of exposure for ambrisentan. In the ARIES E open label long term extension study, the 2 year risk of developing serum aminotransferase elevations >3xULN in patients treated with ambrisentan was 3.9%.
Other clinical information: An improvement in haemodynamic parameters was observed in patients with PAH after 12 weeks (n=29) in a Phase 2 study (AMB220). Treatment with ambrisentan resulted in an increase in mean cardiac index, a decrease in mean pulmonary artery pressure, and a decrease in mean pulmonary vascular resistance.
Decrease in systolic and diastolic blood pressures has been reported with ambrisentan therapy. In placebo controlled clinical trials of 12 weeks duration mean reduction in systolic and diastolic blood pressures from base line to end of treatment were 3 mm Hg and 4.2 mm Hg respectively. The mean decreases in systolic and diastolic blood pressures persisted for up to 4 years of treatment with ambrisentan in the long term open label ARIES E study.
No clinically meaningful effects on the pharmacokinetics of ambrisentan or sildenafil were seen during a drug-drug interaction study in healthy volunteers, and the combination was well tolerated. The number of patients who received concomitant ambrisentan and sildenafil in ARIES-E and AMB222 was 22 patients (5.7%) and 17 patients (47%), respectively. No additional safety concerns were identified in these patients.
Idiopathic Pulmonary Fibrosis: A study of 492 patients (ambrisentan N=329, placebo N=163) with idiopathic pulmonary fibrosis (IPF), 11% of which had secondary pulmonary hypertension (WHO group 3), has been conducted, but was terminated early when it was determined that the primary efficacy endpoint could not be met (ARTEMIS-IPF study). Ninety events (27%) of IPF progression (including respiratory hospitalisations) or death were observed in the ambrisentan group compared to 28 events (17%) in the placebo group. Ambrisentan is therefore contraindicated for patients with IPF with or without secondary pulmonary hypertension (see Contraindications).
Pharmacokinetics: Absorption: Ambrisentan is absorbed rapidly in humans. After oral administration, maximum plasma concentrations (Cmax) of ambrisentan typically occur around 1.5 hours post-dose under both fasted and fed conditions. Cmax and area under the plasma concentration-time curve (AUC) increase dose proportionally over the therapeutic dose range. Steady-state is generally achieved following 4 days of repeat dosing.
A food-effect study involving administration of ambrisentan to healthy volunteers under fasting conditions and with a high-fat meal indicated that the Cmax was decreased 12% while the AUC remained unchanged. This decrease in peak concentration is not clinically significant, and therefore ambrisentan can be taken with or without food.
Distribution: Ambrisentan is highly plasma protein bound. The in vitro plasma protein binding of ambrisentan was, on average, 98.8% and independent of concentration over the range of 0.2 - 20 microgram/ml. Ambrisentan is primarily bound to albumin (96.5%) and to a lesser extent to alpha1-acid glycoprotein.
The distribution of ambrisentan into red blood cells is low, with a mean blood:plasma ratio of 0.57 and 0.61 in males and females, respectively.
Biotransformation: Ambrisentan is a non-sulphonamide (propanoic acid) ERA.
Ambrisentan is glucuronidated via several UGT isoenzymes (UGT1A9S, UGT2B7S and UGT1A3S) to form ambrisentan glucuronide (13%). Ambrisentan also undergoes oxidative metabolism mainly by CYP3A4 and to a lesser extent by CYP3A5 and CYP2C19 to form 4-hydroxymethyl ambrisentan (21%) which is further glucuronidated to 4-hydroxymethyl ambrisentan glucuronide (5%). The binding affinity of 4-hydroxymethyl ambrisentan for the human endothelin receptor is 65-fold less than ambrisentan. Therefore at concentrations observed in the plasma (approximately 4% relative to parent ambrisentan), 4-hydroxymethyl ambrisentan is not expected to contribute to pharmacological activity of ambrisentan.
In vitro data indicate that ambrisentan at 300 μM resulted in less than 50 % inhibition of UGT1A1, UGT1A6, UGT1A9, UGT2B7 (up to 30%) or of cytochrome P450 enzymes 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 and 3A4 (up to 25%). In vitro, ambrisentan has no inhibitory effect on human transporters at clinically relevant concentrations, including Pgp, BCRP, MRP2, BSEP, OATP1B1, OATP1B3 and NTCP. Furthermore, ambrisentan did not induce MRP2, Pgp or BSEP protein expression in rat hepatocytes. Taken together, the in vitro data suggest ambrisentan at clinically relevant concentrations (plasma Cmax up to 3.2 μM) would not be expected to have an effect on UGT1A1, UGT1A6, UGT1A9, UGT2B7 or cytochrome P450 enzymes 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4 or transport via BSEP, BCRP, Pgp, MRP2, OATP1B1/3, or NTCP. The effects of steady-state ambrisentan (10 mg once daily) on the pharmacokinetics and pharmacodynamics of a single dose of warfarin (25 mg), as measured by PT and INR, were investigated in 20 healthy volunteers. Ambrisentan did not have any clinically relevant effects on the pharmacokinetics or pharmacodynamics of warfarin. Similarly, co-administration with warfarin did not affect the pharmacokinetics of ambrisentan (see Interactions).
The effect of 7-day dosing of sildenafil (20 mg three times daily) on the pharmacokinetics of a single dose of ambrisentan, and the effects of 7-day dosing of ambrisentan (10 mg once daily) on the pharmacokinetics of a single dose of sildenafil were investigated in 19 healthy volunteers. With the exception of a 13% increase in sildenafil Cmax following co-administration with ambrisentan, there were no other changes in the pharmacokinetic parameters of sildenafil, N-desmethyl-sildenafil and ambrisentan. This slight increase in sildenafil Cmax is not considered clinically relevant (see Interactions).
The effects of steady-state ambrisentan (10 mg once daily) on the pharmacokinetics of a single dose of tadalafil, and the effects of steady-state tadalafil (40 mg once daily) on the pharmacokinetics of a single dose of ambrisentan were studied in 23 healthy volunteers. Ambrisentan did not have any clinically relevant effects on the pharmacokinetics of tadalafil. Similarly, co-administration with tadalafil did not affect the pharmacokinetics of ambrisentan (see Interactions).
The effects of repeat dosing of ketoconazole (400 mg once daily) on the pharmacokinetics of a single dose of 10 mg ambrisentan were investigated in 16 healthy volunteers. Exposures of ambrisentan as measured by AUC(0-inf) and Cmax were increased by 35% and 20%, respectively. This change in exposure is unlikely to be of any clinical relevance and therefore ambrisentan may be co-administered with ketoconazole.
The effects of repeat dosing of cyclosporine A (100 - 150 mg twice daily) on the steady-state pharmacokinetics of ambrisentan (5 mg once daily), and the effects of repeat dosing of ambrisentan (5 mg once daily) on the steady-state pharmacokinetics of cyclosporine A (100 - 150 mg twice daily) were studied in healthy volunteers. The Cmax and AUC(0-τ) of ambrisentan increased (48% and 121%, respectively) in the presence of multiple doses of cyclosporine A. Based on these changes, the dose of ambrisentan should be limited to 5 mg once daily when co-administered with cyclosporine A (see Dosage & Administration). However, multiple doses of ambrisentan had no clinically relevant effect on cyclosporine A exposure, and no dose adjustment of cyclosporine A is warranted.
The effects of acute and repeat dosing of rifampicin (600 mg once daily) on the steady-state pharmacokinetics of ambrisentan (10 mg once daily) were studied in healthy volunteers. Following initial doses of rifampicin, a transient increase in ambrisentan AUC(0-τ) (121% and 116% after first and second doses of rifampicin, respectively) was observed, presumably due to a rifampicin-mediated OATP inhibition. However, there was no clinically relevant effect on ambrisentan exposure by day 8, following administration of multiple doses of rifampicin. Patients on ambrisentan therapy should be closely monitored when starting treatment with rifampicin (see Precautions and Interactions).
The effects of repeat dosing of ambrisentan (10 mg) on the pharmacokinetics of single dose digoxin were studied in 15 healthy volunteers. Multiple doses of ambrisentan resulted in slight increases in digoxin AUC0-last and trough concentrations, and a 29% increase in digoxin Cmax. The increase in digoxin exposure observed in the presence of multiple doses of ambrisentan was not considered clinically relevant, and no dose adjustment of digoxin is warranted (see Interactions).
The effects of 12 days dosing with ambrisentan (10 mg once daily) on the pharmacokinetics of a single dose of oral contraceptive containing ethinyl estradiol (35 μg) and norethindrone (1 mg) were studied in healthy female volunteers. The Cmax and AUC(0-∞) were slightly decreased for ethinyl estradiol (8% and 4%, respectively), and slightly increased for norethindrone (13% and 14%, respectively). These changes in exposure to ethinyl estradiol or norethindrone were small and are unlikely to be clinically significant (see Interactions).
Elimination: Ambrisentan and its metabolites are eliminated primarily in the bile following hepatic and/or extra-hepatic metabolism. Approximately 22% of the administered dose is recovered in the urine following oral administration with 3.3% being unchanged ambrisentan. Plasma elimination half-life in humans ranges from 13.6 to 16.5 hours.
Special populations: Based on the results of a population pharmacokinetic analysis in healthy volunteers and patients with PAH, the pharmacokinetics of ambrisentan were not significantly influenced by gender or age (see Dosage & Administration).
Renal impairment: Ambrisentan does not undergo significant renal metabolism or renal clearance (excretion). In a population pharmacokinetic analysis, creatinine clearance was found to be a statistically significant covariate affecting the oral clearance of ambrisentan. The magnitude of the decrease in oral clearance is modest (20-40%) in patients with moderate renal impairment and therefore is unlikely to be of any clinical relevance. However, caution should be used in patients with severe renal impairment (see Dosage & Administration).
Hepatic impairment: The main routes of metabolism of ambrisentan are glucuronidation and oxidation with subsequent elimination in the bile and therefore hepatic impairment might be expected to increase exposure (Cmax and AUC) of ambrisentan. In a population pharmacokinetic analysis, the oral clearance was shown to be decreased as a function of increasing bilirubin levels. However, the magnitude of effect of bilirubin is modest (compared to the typical patient with a bilirubin of 0.6 mg/dl, a patient with an elevated bilirubin of 4.5 mg/dl would have approximately 30% lower oral clearance of ambrisentan). The pharmacokinetics of ambrisentan in patients with hepatic impairment (with or without cirrhosis) has not been studied. Therefore ambrisentan should not be initiated in patients with severe hepatic impairment or clinically significant elevated hepatic aminotransferases (>3xULN) (see Contraindications and Precautions).
Toxicology: Preclinical safety data: Due to the class primary pharmacologic effect, a large single dose of ambrisentan (i.e. an overdose) could lower arterial pressure and have the potential for causing hypotension and symptoms related to vasodilation.
Ambrisentan was not shown to be an inhibitor of bile acid transport or to produce overt hepatotoxicity.
Inflammation and changes in the nasal cavity epithelium have been seen in rodents after chronic administration at exposures below the therapeutic levels in humans. In dogs, slight inflammatory responses were observed following chronic high dose administration of ambrisentan at exposures greater than 20-fold that observed in patients.
Nasal bone hyperplasia of the ethmoid turbinates has been observed in the nasal cavity of rats treated with ambrisentan, at exposure levels 3-fold the clinical AUC. Nasal bone hyperplasia has not been observed with ambrisentan in mice or dogs. In the rat, hyperplasia of nasal turbinate bone is a recognised response to nasal inflammation, based on experience with other compounds.
Ambrisentan was clastogenic when tested at high concentrations in mammalian cells in vitro. No evidence for mutagenic or genotoxic effects of ambrisentan were seen in bacteria or in two in vivo rodent studies.
There was no evidence of carcinogenic potential in 2 year oral studies in rats and mice. There was a small increase in mammary fibroadenomas, a benign tumor, in male rats at the highest dose only. Systemic exposure to ambrisentan in male rats at this dose (based on steady-state AUC) was 6-fold that achieved at the 10 mg/day clinical dose.
Testicular tubular atrophy, which was occasionally associated with aspermia, was observed in oral repeat dose toxicity and fertility studies with male rats and mice without safety margin. The testicular changes were not fully recoverable during the off-dose periods evaluated. However no testicular changes were observed in dog studies of up to 39 weeks duration at an exposure 35-fold that seen in humans based on AUC. In male rats, there were no effects of ambrisentan on sperm motility at all doses tested (up to 300 mg/kg/day). A slight (<10%) decrease in the percentage of morphologically normal sperms was noted at 300 mg/kg/day but not at 100 mg/kg/day (>9-fold clinical exposure at 10 mg/day). The effect of ambrisentan on male human fertility is not known.
Ambrisentan has been shown to be teratogenic in rats and rabbits. Abnormalities of the lower jaw, tongue, and/or palate were seen at all doses tested. In addition, the rat study showed an increased incidence of interventricular septal defects, trunk vessel defects, thyroid and thymus abnormalities, ossification of the basisphenoid bone and the occurrence of the umbilical artery located on the left side of the urinary bladder instead of the right side. Teratogenicity is a suspected class effect of ERAs.
Administration of ambrisentan to female rats from late-pregnancy through lactation caused adverse events on maternal behaviour, reduced pup survival and impairment of the reproductive capability of the offspring (with observation of small testes at necropsy), at exposure 3-fold the AUC at the maximum recommended human dose.
In juvenile rats administered ambrisentan orally once daily during postnatal day 7 to 26, 36 or 62, a decrease in brain weight (-3% to -8%) with no morphologic or neurobehavioral changes occurred after breathing sounds, apnoea and hypoxia were observed. These effects occurred at exposures approximately 1.8 to 7 times human paediatric exposures at 10 mg (age 9 to 15 years), based on AUC. The clinical relevance of this finding to the paediatric population is not fully understood.
Volibris is indicated for the treatment of adult patients with pulmonary arterial hypertension (PAH) classified as WHO functional class II and III, to improve exercise capacity (see Pharmacology: Pharmacodynamics under Actions). Efficacy has been shown in idiopathic PAH (IPAH) and in PAH associated with connective tissue disease.
Treatment must be initiated by a physician experienced in the treatment of PAH.
Posology: Volibris is to be taken orally at a dose of 5 mg once daily and may be increased to 10 mg daily depending upon clinical response and tolerability.
In the AMBITION study, patients received 5 mg ambrisentan daily for the first 8 weeks before up titrating to 10 mg, dependent on tolerability (see Pharmacology: Pharmacodynamics under Actions).
Limited data suggest that the abrupt discontinuation of ambrisentan is not associated with rebound worsening of PAH.
When co-administered with cyclosporine A, the dose of ambrisentan should be limited to 5 mg once daily and the patient should be carefully monitored (see Interactions and Pharmacology: Pharmacokinetics under Actions).
Special populations: Elderly patients: No dose adjustment is required in patients over the age of 65 (see Pharmacology: Pharmacokinetics under Actions).
Patients with renal impairment: No dose adjustment is required in patients with renal impairment (see Pharmacology: Pharmacokinetics under Actions). There is limited experience with ambrisentan in individuals with severe renal impairment (creatinine clearance <30 ml/min); therapy should be initiated cautiously in this subgroup and particular care taken if the dose is increased to 10 mg ambrisentan.
Patients with hepatic impairment: Ambrisentan has not been studied in individuals with hepatic impairment (with or without cirrhosis). Since the main routes of metabolism of ambrisentan are glucuronidation and oxidation with subsequent elimination in the bile, hepatic impairment might be expected to increase exposure (Cmax and AUC) to ambrisentan. Therefore ambrisentan must not be initiated in patients with severe hepatic impairment, or clinically significant elevated hepatic aminotransferases (greater than 3 times the Upper Limit of Normal (>3xULN); see Contraindications and Precautions).
Paediatric population: The safety and efficacy of ambrisentan in children and adolescents aged below 18 years has not been established. No clinical data are available (see Pharmacology: Toxicology: Preclinical safety data under Actions regarding data available in juvenile animals).
Method of administration: It is recommended that the tablet is swallowed whole and it can be taken with or without food. It is recommended that the tablet should not be split, crushed or chewed.
There is no experience in PAH patients of ambrisentan at daily doses greater than 10 mg. In healthy volunteers, single doses of 50 and 100 mg (5 to 10 times the maximum recommended dose) were associated with headache, flushing, dizziness, nausea and nasal congestion.
Due to the mechanism of action, an overdose of ambrisentan could potentially result in hypotension (see Pharmacology: Toxicology: Preclinical safety data under Actions). In the case of pronounced hypotension, active cardiovascular support may be required. No specific antidote is available.
Hypersensitivity to the active substance, to soya, or to any of the excipients listed in Description.
Pregnancy (see Use in Pregnancy & Lactation).
Women of child-bearing potential who are not using reliable contraception (see Precautions and Use in Pregnancy & Lactation).
Breast-feeding (see Use in Pregnancy & Lactation).
Severe hepatic impairment (with or without cirrhosis) (see Dosage & Administration).
Baseline values of hepatic aminotransferases (aspartate aminotransferases (AST) and/or alanine aminotransferases (ALT)) >3xULN (see Dosage & Administration and Precautions).
Idiopathic pulmonary fibrosis (IPF), with or without secondary pulmonary hypertension (see Pharmacology: Pharmacodynamics under Actions).
Ambrisentan has not been studied in a sufficient number of patients to establish the benefit/risk balance in WHO functional class I PAH.
The efficacy of ambrisentan as monotherapy has not been established in patients with WHO functional class IV PAH. Therapy that is recommended at the severe stage of the disease (e.g. epoprostenol) should be considered if the clinical condition deteriorates.
Liver function: Liver function abnormalities have been associated with PAH. Cases consistent with autoimmune hepatitis, including possible exacerbation of underlying autoimmune hepatitis, hepatic injury and hepatic enzyme elevations potentially related to therapy have been observed with ambrisentan (see Adverse Reactions and Pharmacology: Pharmacodynamics under Actions). Therefore hepatic aminotransferases (ALT and AST) should be evaluated prior to initiation of ambrisentan and treatment should not be initiated in patients with baseline values of ALT and/or AST>3xULN (see Contraindications).
Patients should be monitored for signs of hepatic injury and monthly monitoring of ALT and AST is recommended. If patients develop sustained, unexplained, clinically significant ALT and/or AST elevation, or if ALT and/or AST elevation is accompanied by signs or symptoms of hepatic injury (e.g. jaundice), ambrisentan therapy should be discontinued.
In patients without clinical symptoms of hepatic injury or of jaundice, re-initiation of ambrisentan may be considered following resolution of hepatic enzyme abnormalities. The advice of a hepatologist is recommended.
Haemoglobin concentration: Reductions in haemoglobin concentrations and haematocrit have been associated with endothelin receptor antagonists (ERAs) including ambrisentan. Most of these decreases were detected during the first 4 weeks of treatment and haemoglobin generally stabilised thereafter. Mean decreases from baseline (ranging from 0.9 to 1.2 g/dL) in haemoglobin concentrations persisted for up to 4 years of treatment with ambrisentan in the long-term open-label extension of the pivotal Phase 3 clinical studies. In the post-marketing period, cases of anaemia requiring blood cell transfusion have been reported (see Adverse Reactions).
Initiation of ambrisentan is not recommended for patients with clinically significant anaemia. It is recommended that haemoglobin and/or haematocrit levels are measured during treatment with ambrisentan, for example at 1 month, 3 months and periodically thereafter in line with clinical practice. If a clinically significant decrease in haemoglobin or haematocrit is observed, and other causes have been excluded, dose reduction or discontinuation of treatment should be considered.
Fluid retention: Peripheral oedema has been observed with ERAs including ambrisentan. Most cases of peripheral oedema in clinical studies with ambrisentan were mild to moderate in severity, although it appeared to occur with greater frequency and severity in patients ≥65 years. Peripheral oedema was reported more frequently with 10 mg ambrisentan in short-term clinical studies (see Adverse Reactions).
Post-marketing reports of fluid retention occurring within weeks after starting ambrisentan have been received and, in some cases, have required intervention with a diuretic or hospitalisation for fluid management or decompensated heart failure. If patients have pre-existing fluid overload, this should be managed as clinically appropriate prior to starting ambrisentan.
If clinically significant fluid retention develops during therapy with ambrisentan, with or without associated weight gain, further evaluation should be undertaken to determine the cause, such as ambrisentan or underlying heart failure, and the possible need for specific treatment or discontinuation of ambrisentan therapy.
Women of child-bearing potential: Volibris treatment must not be initiated in women of child-bearing potential unless the result of a pre-treatment pregnancy test is negative and reliable contraception is practiced. If there is any doubt on what contraceptive advice should be given to the individual patient, consultation with a gynaecologist should be considered. Monthly pregnancy tests during treatment with ambrisentan are recommended (see Contraindications and Use in Pregnancy & Lactation).
Pulmonary veno-occlusive disease: Cases of pulmonary oedema have been reported with vasodilating medicinal products, such as ERAs, when used in patients with pulmonary veno-occlusive disease. Consequently, if PAH patients develop acute pulmonary oedema when treated with ambrisentan, the possibility of pulmonary veno-occlusive disease should be considered.
Concomitant use with other medicinal products: Patients on ambrisentan therapy should be closely monitored when starting treatment with rifampicin (see Interactions and Pharmacology: Pharmacokinetics under Actions).
Excipients: Volibris tablets contain lactose. Patients with rare hereditary problems of galactose intolerance, the Lapp lactase deficiency or glucose-galactose malabsorption should not take this medicine.
Volibris tablets contain the azo colouring agent Allura red AC Aluminium Lake (E129), which can cause allergic reactions.
Volibris tablets contain lecithin derived from soya. If a patient is hypersensitive to soya, ambrisentan must not be used (see Contraindications).
Volibris tablets contain less than 1 mmol sodium (23 mg), which is essentially 'sodium-free'.
Effects on ability to drive and use machines: Ambrisentan has minor or moderate influence on the ability to drive and use machines. The clinical status of the patient and the adverse reaction profile of ambrisentan (such as hypotension, dizziness, asthenia, fatigue) should be borne in mind when considering the patient's ability to perform tasks that require judgement, motor or cognitive skills (see Adverse Reactions). Patients should be aware of how they might be affected by ambrisentan before driving or using machines.
Women of childbearing potential: Ambrisentan treatment must not be initiated in women of child-bearing potential unless the result of a pre-treatment pregnancy test is negative and reliable contraception is practiced. Monthly pregnancy tests during treatment with ambrisentan are recommended.
Pregnancy: Ambrisentan is contraindicated in pregnancy (see Contraindications). Animal studies have shown that ambrisentan is teratogenic. There is no experience in humans.
Women receiving ambrisentan must be advised of the risk of foetal harm and alternative therapy initiated if pregnancy occurs (see Contraindications, Precautions and Pharmacology: Toxicology: Preclinical safety data under Actions).
Breast-feeding: It is not known whether ambrisentan is excreted in human breast milk. The excretion of ambrisentan in milk has not been studied in animals. Therefore breast-feeding is contraindicated in patients taking ambrisentan (see Contraindications).
Male fertility: The development of testicular tubular atrophy in male animals has been linked to the chronic administration of ERAs, including ambrisentan (see Pharmacology: Toxicology: Preclinical safety data under Actions). Although no clear evidence of a detrimental effect of ambrisentan long-term exposure on sperm count was found in ARIES-E study, chronic administration of ambrisentan was associated with changes in markers of spermatogenesis. A decrease in plasma inhibin-B concentration and an increase in plasma FSH concentration were observed. The effect on male human fertility is not known but a deterioration of spermatogenesis cannot be excluded. Chronic administration of ambrisentan was not associated with a change in plasma testosterone in clinical studies.
Summary of the safety profile: The safety of ambrisentan has been evaluated as monotherapy and/or in combination in clinical trials of more than 1200 patients with PAH (see Pharmacology: Pharmacodynamics under Actions). Adverse reactions identified from 12 week placebo controlled clinical trial data are included as follows by system organ class and frequency. Information from longer term non-placebo controlled studies (ARIES-E and AMBITION) is also included as follows. No previously unknown adverse reactions were identified with long-term treatment. With longer observation in uncontrolled studies (mean observation of 79 weeks), the safety profile was similar to that observed in the short term studies. Routine pharmacovigilance data are also presented.
Peripheral oedema, fluid retention and headache (including sinus headache, migraine) were the most common adverse reactions observed with ambrisentan. The higher dose (10 mg) was associated with a higher incidence of these adverse reactions, and peripheral oedema tended to be more severe in patients ≥65 years in short-term clinical studies (see Precautions).
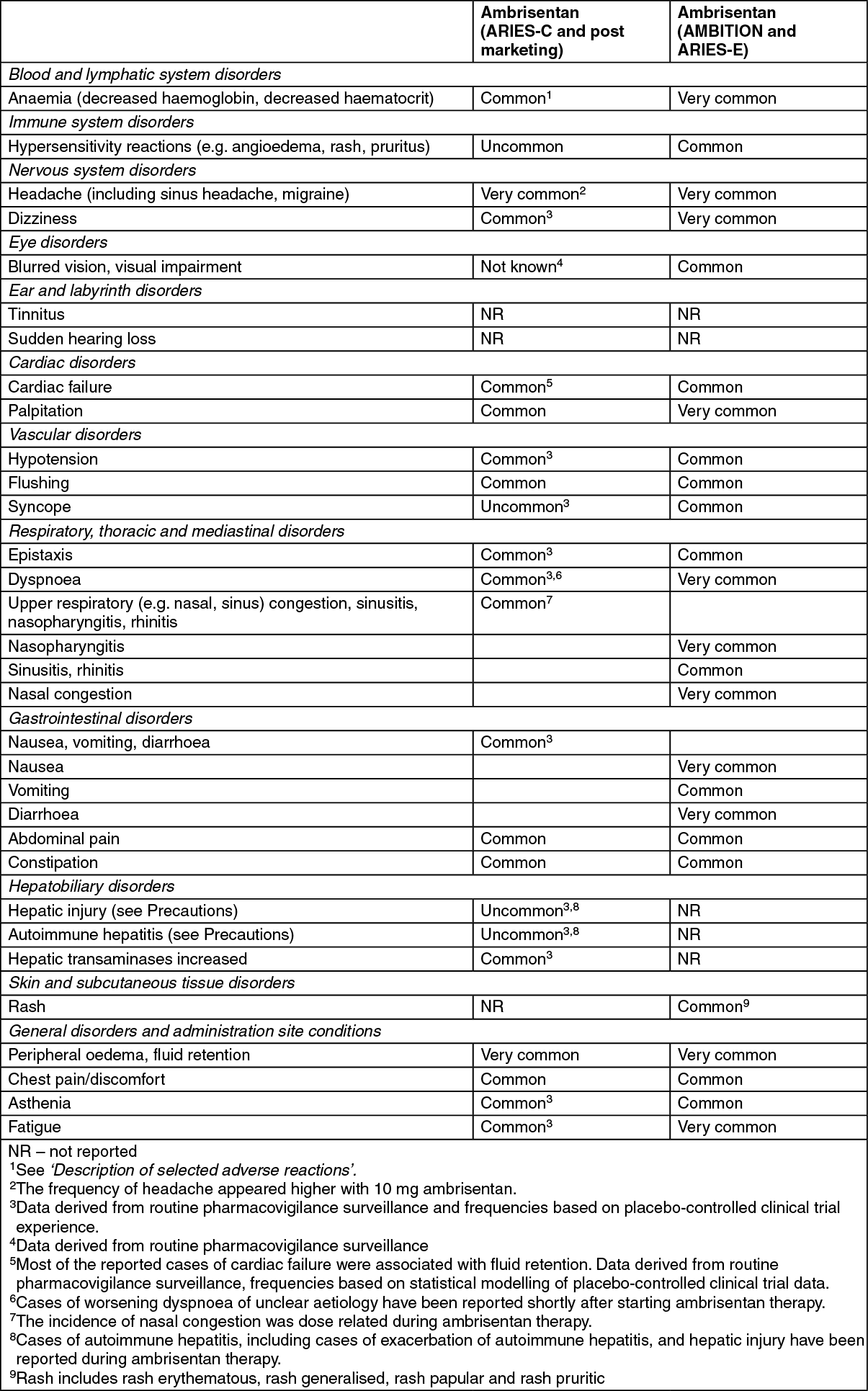
Tabulated list of adverse reactions: Frequencies are defined as: very common (≥ 1/10); common (≥ 1/100 to <1/10); uncommon (≥1/1,000 to <1/100); rare (≥1/10,000 to <1/1,000); very rare (<1/10,000) and not known (cannot be estimated from available data). For dose-related adverse reactions the frequency category reflects the higher dose of ambrisentan. Frequency categories do not account for other factors including varying study duration, pre-existing conditions and baseline patient characteristics. Adverse reaction frequency categories assigned based on clinical trial experience may not reflect the frequency of adverse events occurring during normal clinical practice. Within each frequency grouping, adverse reactions are presented in order of decreasing seriousness. (See table.)
 Click on icon to see table/diagram/image
Description of selected adverse reactions: Decreased haemoglobin:
Click on icon to see table/diagram/image
Description of selected adverse reactions: Decreased haemoglobin: In the post-marketing period, cases of anaemia requiring blood cell transfusion have been reported (see Precautions). The frequency of decreased haemoglobin (anaemia) was higher with 10 mg ambrisentan. Across the 12 week placebo controlled Phase 3 clinical studies, mean haemoglobin concentrations decreased for patients in the ambrisentan groups and were detected as early as week 4 (decrease by 0.83 g/dL); mean changes from baseline appeared to stabilise over the subsequent 8 weeks. A total of 17 patients (6.5%) in the ambrisentan treatment groups had decreases in haemoglobin of ≥15% from baseline and which fell below the lower limit of normal.
Ambrisentan does not inhibit or induce phase I or II drug metabolising enzymes at clinically relevant concentrations in in vitro and in vivo non-clinical studies, suggesting a low potential for ambrisentan to alter the profile of medicinal products metabolised by these pathways.
The potential for ambrisentan to induce CYP3A4 activity was explored in healthy volunteers with results suggesting a lack of inductive effect of ambrisentan on the CYP3A4 isoenzyme.
Cyclosporine A: Steady-state co-administration of ambrisentan and cyclosporine A resulted in a 2-fold increase in ambrisentan exposure in healthy volunteers. This may be due to the inhibition by cyclosporine A of transporters and metabolic enzymes involved in the pharmacokinetics of ambrisentan. Therefore the dose of ambrisentan should be limited to 5 mg once daily when co-administered with cyclosporine A (see Dosage & Administration). Multiple doses of ambrisentan had no effect on cyclosporine A exposure, and no dose adjustment of cyclosporine A is warranted.
Rifampicin: Co-administration of rifampicin (an inhibitor of Organic Anion Transporting Polypeptide [OATP], a strong inducer of CYP3A and 2C19, and inducer of P-gp and uridine-diphospho-glucuronosyltransferases [UGTs]) was associated with a transient (approximately 2-fold) increase in ambrisentan exposure following initial doses in healthy volunteers. However, by day 8, steady state administration of rifampicin had no clinically relevant effect on ambrisentan exposure. Patients on ambrisentan therapy should be closely monitored when starting treatment with rifampicin (see Precautions and Pharmacology: Pharmacokinetics under Actions).
Other targeted PAH treatments: The efficacy and safety of ambrisentan when co-administered with other treatments for PAH (e.g. prostanoids and soluble guanylate cyclase stimulators) has not been specifically studied in controlled clinical trials in PAH patients (see Pharmacology: Pharmacodynamics under Actions). No specific drug-drug interactions with soluble guanylate cyclase stimulators or prostanoids are anticipated based on the known biotransformation data (see Pharmacology: Pharmacokinetics under Actions). However, no specific drug-drug interactions studies have been conducted with these drugs. Therefore, caution is recommended in the case of co-administration.
Phosphodiesterase inhibitors: Co-administration of ambrisentan with a phosphodiesterase inhibitor, either sildenafil or tadalafil (both substrates of CYP3A4) in healthy volunteers did not significantly affect the pharmacokinetics of the phosphodiesterase inhibitor or ambrisentan (see Pharmacology: Pharmacokinetics under Actions).
Oral contraceptives: In a clinical study in healthy volunteers, steady-state dosing with ambrisentan 10 mg once daily did not significantly affect the single-dose pharmacokinetics of the ethinyl estradiol and norethindrone components of a combined oral contraceptive (see Pharmacology: Pharmacokinetics under Actions). Based on this pharmacokinetic study, ambrisentan would not be expected to significantly affect exposure to oestrogen- or progestogen- based contraceptives.
Warfarin: Ambrisentan had no effects on the steady-state pharmacokinetics and anti-coagulant activity of warfarin in a healthy volunteer study (see Pharmacology: Pharmacokinetics under Actions). Warfarin also had no clinically significant effects on the pharmacokinetics of ambrisentan. In addition, in patients, ambrisentan had no overall effect on the weekly warfarin-type anticoagulant dose, prothrombin time (PT) and international normalised ratio (INR).
Ketoconazole: Steady-state administration of ketoconazole (a strong inhibitor of CYP3A4) did not result in a clinically significant increase in exposure to ambrisentan (see Pharmacology: Pharmacokinetics under Actions).
Effect of ambrisentan on xenobiotic transporters: In vitro, ambrisentan has no inhibitory effect on human transporters at clinically relevant concentrations, including the P-glycoprotein (Pgp), breast cancer resistance protein (BCRP), multi-drug resistance related protein 2 (MRP2), bile salt export pump (BSEP), organic anion transporting polypeptides (OATP1B1 and OATP1B3) and the sodium-dependent taurocholate co-transporting polypeptide (NTCP).
Ambrisentan is a substrate for Pgp-mediated efflux.
In vitro studies in rat hepatocytes also showed that ambrisentan did not induce Pgp, BSEP or MRP2 protein expression.
Steady-state administration of ambrisentan in healthy volunteers had no clinically relevant effects on the single-dose pharmacokinetics of digoxin, a substrate for Pgp (see Pharmacology: Pharmacokinetics under Actions).
Special precautions for disposal: No special requirements for disposal.
Incompatibilities: Not applicable.
C02KX02 - ambrisentan ; Belongs to the class of other antihypertensives. Used in the treatment of pulmonary arterial hypertension.
Volibris FC tab 10 mg
30's
Volibris FC tab 5 mg
30's


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out
