Pharmacotherapeutic group: Antineoplastic agents, protein kinase inhibitors, B-Raf serine-threonine kinase (BRAF) inhibitors.
ATC code: L01EC02.
Pharmacology: Pharmacodynamics: Mechanism of action: Dabrafenib is an inhibitor of RAF kinases. Oncogenic mutations in BRAF lead to constitutive activation of the RAS/RAF/MEK/ERK pathway. BRAF mutations have been identified at a high frequency in specific cancers, including approximately 50% of melanoma. The most commonly observed BRAF mutation is V600E which accounts for approximately 90% of the BRAF mutations that are seen in melanoma.
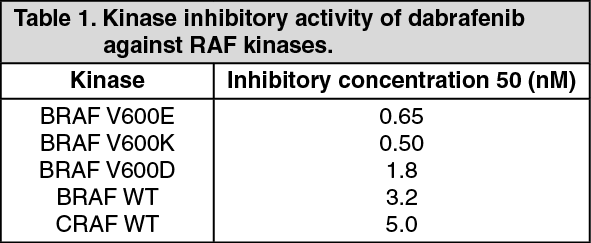
Preclinical data generated in biochemical assays demonstrated that dabrafenib inhibits BRAF kinases with activating codon 600 mutations (see Table 1).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Dabrafenib demonstrated suppression of a downstream pharmacodynamic biomarker (phosphorylated ERK) and inhibited cell growth of BRAF V600 mutant melanoma cell lines,
in vitro and in animal models.
In subjects with BRAF V600 mutation positive melanoma, administration of dabrafenib resulted in inhibition of tumour phosphorylated ERK relative to baseline.
Combination with trametinib: Trametinib is a reversible, highly selective, allosteric inhibitor of mitogen-activated extracellular signal regulated kinase 1 (MEK1) and MEK2 activation and kinase activity. MEK proteins are components of the extracellular signal-related kinase (ERK) pathway. Thus, trametinib and dabrafenib inhibit two kinases in this pathway, MEK and RAF, and therefore the combination provides concomitant inhibition of the pathway. The combination of dabrafenib with trametinib has shown anti-tumour activity in BRAF V600 mutation positive melanoma cell lines
in vitro and delays the emergence of resistance
in vivo in BRAF V600 mutation positive melanoma xenografts.
Determination of BRAF mutation status: Before taking dabrafenib or combination with trametinib, patients must have BRAF V600 mutation-positive tumour status confirmed by a validated test. In the Phase II and III clinical trials, screening for eligibility required central testing for BRAF V600 mutation using a BRAF mutation assay conducted on the most recent tumour sample available. Primary tumour or tumour from a metastatic site was tested with an investigational use only assay (IUO). The IUO is an allele-specific polymerase chain reaction (PCR) assay performed on DNA extracted from formalin-fixed paraffin-embedded (FFPE) tumour tissue. The assay was specifically designed to differentiate between the V600E and V600K mutations. Only subjects with BRAF V600E or V600K mutation positive tumours were eligible for study participation.
Subsequently, all patient samples were re-tested using the bioMerieux (bMx) THxID BRAF validated assay, which has CE marking. The bMx THxID BRAF assay is an allele-specific PCR performed on DNA extracted from FFPE tumour tissue. The assay was designed to detect the BRAF V600E and V600K mutations with high sensitivity (down to 5% V600E and V600K sequence in a background of wild-type sequence using DNA extracted from FFPE tissue). Non-clinical and clinical trials with retrospective bi-directional Sanger sequencing analyses have shown that the test also detects the less common BRAF V600D mutation and V600E/K601E mutation with lower sensitivity. Of the specimens from the non-clinical and clinical trials (n=876) that were mutation positive by the THxID BRAF assay and subsequently were sequenced using the reference method, the specificity of the assay was 94%.
Clinical efficacy and safety: Unresectable or metastatic melanoma: Dabrafenib in combination with trametinib: Treatment-naïve patients: The efficacy and safety of the recommended dose of trametinib (2 mg once daily) in combination with dabrafenib (150 mg twice daily) for the treatment of adult patients with unresectable or metastatic melanoma with a BRAF V600 mutation was studied in two Phase III trials and one supportive Phase I/II study.
MEK115306 (COMBI-d): MEK115306 was a Phase III, randomised, double-blinded study comparing the combination of dabrafenib and trametinib to dabrafenib and placebo in first-line therapy for subjects with unresectable (Stage IIIC) or metastatic (Stage IV) BRAF V600E/K mutation-positive cutaneous melanoma. The primary endpoint of the study was progression-free survival (PFS), with a key secondary endpoint of overall survival (OS). Subjects were stratified by lactate dehydrogenase (LDH) level (> the upper limit of normal (ULN) versus ≤ULN) and BRAF mutation (V600E versus V600K).
A total of 423 subjects were randomised 1:1 to either combination (N=211) or dabrafenib (N=212). Most subjects were Caucasian (>99%) and male (53%), with a median age of 56 years (28% were ≥65 years). The majority of subjects had Stage IVM1c disease (67%). Most subjects had LDH ≤ULN (65%), Eastern Cooperative Oncology Group (ECOG) performance status of 0 (72%), and visceral disease (73%) at baseline. The majority of subjects had a BRAF V600E mutation (85%). Subjects with brain metastases were not included in the trial.
Median OS and estimated 1-year, 2-year, 3-year, 4 year and 5-year survival rates are presented in Table 2. From an OS analysis at 5 years, the median OS for the combination arm was approximately 7 months longer than for dabrafenib monotherapy (25.8 months versus 18.7 months) with 5-year
survival rates of 32% for the combination versus 27% for dabrafenib monotherapy (Table 2, Figure 1). The Kaplan-Meier OS curve appears to stabilise from 3 to 5 years (see Figure 1). The 5-year overall survival rate was 40% (95% CI: 31.2, 48.4) in the combination arm versus 33% (95% CI: 25.0, 41.0) in the dabrafenib monotherapy arm for patients who had a normal lactate dehydrogenase level at baseline, and 16% (95% CI: 8.4, 26.0) in the combination arm versus 14% (95% CI: 6.8, 23.1) in the dabrafenib monotherapy arm for patients with an elevated lactate dehydrogenase level at baseline. (See Table 2 and Figure 1.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Improvements for the primary endpoint of PFS were sustained over a 5-year timeframe in the combination arm compared to dabrafenib monotherapy. Improvements were also observed for overall response rate (ORR) and a longer duration of response (DoR) was observed in the combination arm compared to dabrafenib monotherapy (see Table 3).
Click on icon to see table/diagram/image
MEK116513 (COMBI-v): Study MEK116513 was a 2-arm, randomised, open-label, Phase III study comparing dabrafenib and trametinib combination therapy with vemurafenib monotherapy in BRAF V600 mutation-positive unresectable or metastatic melanoma. The primary endpoint of the study was OS with a key secondary endpoint of PFS. Subjects were stratified by lactate dehydrogenase (LDH) level (> the upper limit of normal (ULN) versus ≤ULN) and BRAF mutation (V600E versus V600K).
A total of 704 subjects were randomised 1:1 to either combination or vemurafenib. Most subjects were Caucasian (>96%) and male (55%), with a median age of 55 years (24% were ≥65 years). The majority of subjects had Stage IV M1c disease (61% overall). Most subjects had LDH ≤ULN (67%), ECOG performance status of 0 (70%), and visceral disease (78%) at Baseline. Overall, 54% of subjects had <3 disease sites at baseline. The majority of subjects had BRAF V600E mutation-positive melanoma (89%). Subjects with brain metastases were not included in the trial.
Median OS and estimated 1-year, 2-year, 3-year, 4 year and 5-year survival rates are presented in Table 4. From an OS analysis at 5 years, the median OS for the combination arm was approximately 8 months longer than the median OS for vemurafenib monotherapy (26.0 months versus 17.8 months) with 5-year survival rates of 36% for the combination versus 23% for vemurafenib monotherapy (Table 4, Figure 2). The Kaplan-Meier OS curve appears to stabilise from 3 to 5 years (see Figure 2). The 5-year overall survival rate was 46% (95% CI: 38.8, 52.0) in the combination arm versus 28% (95% CI: 22.5, 34.6) in the vemurafenib monotherapy arm for patients who had a normal lactate dehydrogenase level at baseline, and 16% (95% CI: 9.3, 23.3) in the combination arm versus 10% (95% CI: 5.1, 17.4) in the vemurafenib monotherapy arm for patients with an elevated lactate dehydrogenase level at baseline. (See Table 4 and Figure 2.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Improvements for the secondary endpoint of PFS were sustained over a 5-year timeframe in the combination arm compared to vemurafenib monotherapy. Improvements were also observed for ORR and a longer DoR was observed in the combination arm compared to vemurafenib monotherapy (see Table 5).
Click on icon to see table/diagram/image
Prior BRAF inhibitor therapy: There are limited data in patients taking the combination of dabrafenib with trametinib who have progressed on a prior BRAF inhibitor.
Part B of study BRF113220 included a cohort of 26 patients that had progressed on a BRAF inhibitor. The trametinib 2 mg once daily and dabrafenib 150 mg twice daily combination demonstrated limited clinical activity in patients who had progressed on a BRAF inhibitor. The investigator-assessed confirmed response rate was 15% (95% CI: 4.4, 34.9) and the median PFS was 3.6 months (95% CI: 1.9, 5.2). Similar results were seen in the 45 patients who crossed over from dabrafenib monotherapy to the trametinib 2 mg once daily and dabrafenib 150 mg twice daily combination in Part C of this study. In these patients a 13% (95 CI: 5.0, 27.0) confirmed response rate was observed with a median PFS of 3.6 months (95% CI: 2, 4).
Patients with brain metastases: The efficacy and safety of dabrafenib in combination with trametinib in patients with BRAF mutant-positive melanoma that has metastasised to the brain was studied in a non-randomised, open-label, multicentre, Phase II study (COMBI-MB study). A total of 125 patients were enrolled into four cohorts: Cohort A: patients with BRAFV600E mutant melanoma with asymptomatic brain metastases without prior local brain-directed therapy and ECOG performance status of 0 or 1.
Cohort B: patients with BRAFV600E mutant melanoma with asymptomatic brain metastases with prior local brain-directed therapy and ECOG performance status of 0 or1.
Cohort C: patients with BRAFV600D/K/R mutant melanoma with asymptomatic brain metastases, with or without prior local brain-directed therapy and ECOG performance status of 0 or 1.
Cohort D: patients with BRAFV600D/E/K/R mutant melanoma with symptomatic brain metastases, with or without prior local brain-directed therapy and ECOG performance status of 0 or 1 or 2.
The primary endpoint of the study was intracranial response in Cohort A, defined as the percentage of patients with a confirmed intracranial response assessed by the investigator using modified Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1. Intracranial response assessed by the investigator in Cohorts B, C and D were secondary endpoints of the study. Due to small sample size reflected by wide 95% CIs, the results in Cohorts B, C, and D should be interpreted with caution. Efficacy results are summarised in Table 6. (See Table 6.)
Click on icon to see table/diagram/image
Dabrafenib monotherapy: The efficacy of dabrafenib in the treatment of adult patients with BRAF V600 mutation positive unresectable or metastatic melanoma has been evaluated in 3 clinical trials (BRF113683 [BREAK-3], BRF113929 [BREAK-MB], and BRF113710 [BREAK-2]) including patients with BRAF V600E and/or V600K mutations.
Included in these clinical trials were in total 402 subjects with BRAF V600E and 49 subjects with BRAF V600K mutation. Patients with melanoma driven by BRAF mutations other than V600E were excluded from the confirmatory trial and with respect to patients with the V600K mutation in single arm clinical trials the activity appears lower than in V600E tumours.
No data is available in patients with melanoma harbouring BRAF V600 mutations others than V600E and V600K. Efficacy of dabrafenib in subjects previously treated with a protein kinase inhibitor has not been investigated.
Previously untreated patients (results from the Phase III study [BREAK-3]): The efficacy and safety of dabrafenib were evaluated in a Phase III randomised, open-label study [BREAK 3] comparing dabrafenib to dacarbazine (DTIC) in previously untreated patients with BRAF V600E mutation positive advanced (unresectable Stage III) or metastatic (Stage IV) melanoma. Patients with melanoma driven by BRAF mutations other than V600E were excluded.
The primary objective for this study was to evaluate the efficacy of dabrafenib compared to DTIC with respect to PFS per investigator assessment. Patients on the DTIC arm were allowed to cross over to dabrafenib after independent radiographic confirmation of initial progression. Baseline characteristics were balanced between treatment groups. Sixty percent of patients were male and 99.6% were Caucasian; the median age was 52 years with 21% of patients being ≥65 years, 98.4% had ECOG status of 0 or 1, and 97% of patients had metastatic disease.
At the pre-specified analysis with a 19 December 2011 data cut, a significant improvement in the primary endpoint of PFS (HR=0.30; 95% CI 0.18, 0.51; p < 0.0001) was achieved. Efficacy results from the primary analysis and a post-hoc analysis with 6-months additional follow up are summarised in Table 7. OS data from a further post-hoc analysis based on a 18 December 2012 data cut are shown in Figure 3. (See Table 7.)
Click on icon to see table/diagram/image
As of 25 June 2012 cut-off, thirty five subjects (55.6%) of the 63 randomised to DTIC had crossed over to dabrafenib and 63% of subjects randomised to dabrafenib and 79% of subjects randomised to DTIC had progressed or died. Median PFS after cross-over was 4.4 months. (See Table 8.)
Click on icon to see table/diagram/image
OS data from a further post-hoc analysis based on the 18 December 2012 data cut demonstrated a 12-month OS rate of 63% and 70% for DTIC and dabrafenib treatments, respectively. (See Figure 3.)
Click on icon to see table/diagram/image
Patients with brain metastases (results from the Phase II study (BREAK-MB): BREAK-MB was a multicentre, open-label, two-cohort, Phase II study designed to evaluate the intracranial response of dabrafenib in subjects with histologically confirmed (Stage IV) BRAF-mutation positive (V600E or V600K) melanoma metastatic to the brain. Subjects were enrolled into Cohort A (subjects with no prior local therapy for brain metastasis) or Cohort B (subjects who received prior local therapy for brain metastasis).
The primary endpoint of the study was overall intracranial response rate (OIRR) in the V600E patient population, as assessed by investigators. The confirmed OIRR and other efficacy results per investigator assessment are presented in Table 9. (See Table 9.)
Click on icon to see table/diagram/image
Patients who were previously untreated or failed at least one prior systemic therapy (results from the Phase II [BREAK-2]): BRF113710 (BREAK-2) was a multicentre, single-arm study that enrolled 92 subjects with metastatic melanoma (Stage IV) with confirmed BRAF V600E or V600K mutation-positive melanoma.
The investigator assessed confirmed response rate in patients with BRAF V600E metastatic melanoma (n=76) was 59% (95% CI: 48.2, 70.3) and the median DoR was 5.2 months (95% CI: 3.9, not calculable) based on a median follow-up time of 6.5 months. In patients with BRAF V600K mutation positive metastatic melanoma (n=16) the response rate was 13% (95% CI: 0.0, 28.7) with a median DoR of 5.3 months (95% CI: 3.7, 6.8). Although limited by the low number of patients, median OS appeared consistent with data in patients with BRAF V600E positive tumours.
Adjuvant treatment of Stage III melanoma: BRF115532 (COMBI-AD): The efficacy and safety of dabrafenib in combination with trametinib were studied in a Phase III, multicentre, randomised, double-blind, placebo-controlled study in patients with Stage III (Stage IIIA [lymph node metastasis >1 mm], IIIB, or IIIC) cutaneous melanoma with a BRAF V600 E/K mutation, following complete resection.
Patients were randomised 1:1 to receive either combination therapy (dabrafenib 150 mg twice daily and trametinib 2 mg once daily) or two placebos for a period of 12 months. Enrollment required complete resection of melanoma with complete lymphadenectomy within 12 weeks prior to randomisation. Any prior systemic anti-cancer treatment, including radiotherapy, was not allowed. Patients with a history of prior malignancy, if disease-free for at least 5 years, were eligible. Patients presenting with malignancies with confirmed activating RAS mutations were not eligible. Patients were stratified by BRAF mutation status (V600E versus V600K) and stage of disease prior to surgery using the American Joint Committee on Cancer (AJCC) 7th edition Melanoma Staging System (by Stage III sub-stage, indicating different levels of lymph node involvement and primary tumour size and ulceration). The primary endpoint was investigator-assessed relapse-free survival (RFS), defined as the time from randomisation to disease recurrence or death from any cause. Radiological tumour assessment was conducted every 3 months for the first two years and every 6 months thereafter, until first relapse was observed. Secondary endpoints include overall survival (OS; key secondary endpoint), freedom from relapse (FFR) and distant metastasis-free survival (DMFS).
A total of 870 patients were randomised to the combination therapy (n=438) and placebo (n=432) arms. Most patients were Caucasian (99%) and male (55%), with a median age of 51 years (18% were ≥65 years). The study included patients with all sub-stages of Stage III disease prior to resection; 18% of these patients had lymph node involvement only identifiable by microscope and no primary tumour ulceration. The majority of patients had a BRAF V600E mutation (91%). At the time of the primary analysis, the median duration of follow-up (time from randomisation to last contact or death) was 2.83 years in the dabrafenib and trametinib combination arm and 2.75 years in the placebo arm
Results for the primary analysis of RFS are presented in Table 10. The study showed a statistically significant difference for the primary outcome of RFS between treatment arms, with a median RFS of 16.6 months for the placebo arm and not yet reached for the combination arm (HR: 0.47; 95% confidence interval: (0.39, 0.58); p=1.53 x 10
-14). The observed RFS benefit was consistently demonstrated across subgroups of patients including age, sex and race. Results were also consistent across stratification factors for disease stage and BRAF V600 mutation type. (See Table 10.)
Click on icon to see table/diagram/image
Based on updated data with an additional 29 months of follow-up compared to the primary analysis (minimum follow-up of 59 months), the RFS benefit was maintained with an estimated HR of 0.51 (95% CI: 0.42, 0.61) (Figure 4). The 5-year RFS rate was 52% (95% CI: 48, 58) in the combination arm compared to 36% (95% CI: 32, 41) in the placebo arm. (See Figure 4.)
Click on icon to see table/diagram/image
Based on 153 events (60 [14%] in the combination arm and 93 [22%] in the placebo arm) corresponding to a 26% information fraction of the total target of 597 OS events, the estimated hazard ratio for OS was 0.57 (95% CI: 0.42, 0.79; p=0.0006). These results did not meet the pre-specified boundary to claim statistical significance at this first OS interim analysis (HR=0.50; p=0.000019). Survival estimates at 1 and 2 years from randomisation were 97% and 91% in the combination arm and 94% and 83% in the placebo arm, respectively).
Non-small cell lung cancer: Study BRF113928: The efficacy and safety of dabrafenib in combination with trametinib was studied in a Phase II, three-cohort, multicentre, non-randomised and open-label study in which patients with stage IV BRAF V600E mutant NSCLC were enrolled. The primary endpoint was ORR using the RECIST 1.1 assessed by the investigator. Secondary endpoints included DoR, PFS, OS, safety and population pharmacokinetics. ORR, DoR and PFS were also assessed by an Independent Review Committee (IRC) as a sensitivity analysis.
Cohorts were enrolled sequentially: Cohort A: Monotherapy (dabrafenib 150 mg twice daily), 84 patients enrolled. 78 patients had previous systemic treatment for their metastatic disease.
Cohort B: Combination therapy (dabrafenib 150 mg twice daily and trametinib 2 mg once daily), 59 patients enrolled. 57 patients had 1-3 lines of previous systemic treatment for their metastatic disease. 2 patients had no previous systemic treatment and were included in the analysis for patients enrolled in Cohort C.
Cohort C: Combination therapy (dabrafenib 150 mg twice daily and trametinib 2 mg once daily), 34 patients. All patients received study medicinal product as first-line treatment for metastatic disease.
Among the total of 93 patients who were enrolled in the combination therapy cohorts B and C, most patients were Caucasian (>90%), and similar female versus male (54% versus 46%), with a median age of 64 years in second line or higher patients and 68 years in the first line patients. Most patients (94%) enrolled in the combination therapy treated cohorts had an ECOG performance status of 0 or 1. 26 (28%) had never smoked. The majority of patients had a non-squamous histology. In the previously treated population, 38 patients (67%) had one line of systemic anti-cancer therapy for metastatic disease.
At the time of the primary analysis, the primary endpoint of investigator-assessed ORR in the first line population was 61.1% (95% CI, 43.5%, 76.9%), and in the previously treated population was 66.7% (95% CI, 52.9%, 78.6%). These met the statistical significance to reject the null hypothesis that the ORR of dabrafenib in combination with trametinib for this NSCLC population was less than or equal to 30%. The ORR results assessed by IRC were consistent with the investigator assessment. The efficacy of the combination with trametinib was superior when indirectly compared to dabrafenib monotherapy in Cohort A. The final analysis of efficacy performed 5 years after last subject first dose is presented in Table 11. (See Table 11.)
Click on icon to see table/diagram/image
QT prolongation: Worst-case QTc prolongation of >60 millisecond (msec) was observed in 3% of dabrafenib-treated subjects (one >500 msec in the integrated safety population). In the Phase III study MEK115306 no patients treated with trametinib in combination with dabrafenib had worst-case QTcB prolongation to >500 msec; QTcB was increased more than 60 msec from baseline in 1% (3/209) of patients. In the Phase III study MEK116513 four patients (1%) treated with trametinib in combination with dabrafenib had a QTcB Grade 3 increase (>500 msec). Two of these patients had a QTcB Grade 3 increase (>500 msec) that was also an increase >60 msec from baseline.
The potential effect of dabrafenib on QT prolongation was assessed in a dedicated multiple dose QT study. A supratherapeutic dose of 300 mg dabrafenib twice daily was administered in 32 subjects with BRAF V600 mutation-positive tumours. No clinically relevant effect of dabrafenib or its metabolites on the QTc interval was observed.
Other studies - pyrexia management analysis: Study CPDR001F2301 (COMBI-i) and Study CDRB436F2410 (COMBI-Aplus): Pyrexia is observed in patients treated with dabrafenib and trametinib combination therapy. The initial registration studies for the combination therapy in the unresectable or metastatic melanoma setting (COMBI-d and COMBI-v; total N=559) and in the adjuvant melanoma setting (COMBI-AD, N=435) recommended to interrupt only dabrafenib in case of pyrexia (fever ≥38.5°C). In two subsequent studies in unresectable or metastatic melanoma (COMBI-i control arm, N=264) and in the adjuvant melanoma setting (COMBI-Aplus, N=552), interruption of both medicinal products when patient's temperature is ≥38°C (COMBI-Aplus), or at the first symptom of pyrexia (COMBI-i; COMBI-Aplus for recurrent pyrexia) was advised. In COMBI-i and COMBI-Aplus there was a lower incidence of grade 3/4 pyrexia, complicated pyrexia, hospitalisation due to serious pyrexia adverse events of special interest (AESIs), the time spent in pyrexia AESIs, and permanent discontinuations from both medicinal products due to pyrexia AESIs (the latter in the adjuvant setting only) compared to COMBI-d, COMBI-v and COMBI-AD. The COMBI-Aplus study met its primary endpoint with a composite rate of 8.0% (95% CI: 5.9, 10.6) for grade 3/4 pyrexia, hospitalisation due to pyrexia, or permanent treatment discontinuation due to pyrexia compared to 20.0% (95% CI: 16.3, 24.1) for the historical control (COMBI-AD).
Paediatric population: The European Medicines Agency has deferred the obligation to submit the results of studies with dabrafenib in one or more subsets of the paediatric population in melanoma and solid malignant tumours (see Dosage & Administration for information on paediatric use).
Pharmacokinetics: Absorption: Dabrafenib is absorbed orally with median time to achieve peak plasma concentration of 2 hours post-dose. Mean absolute bioavailability of oral dabrafenib is 95% (90% CI: 81, 110%). Dabrafenib exposure (C
max and AUC) increased in a dose proportional manner between 12 and 300 mg following single-dose administration, but the increase was less than dose-proportional after repeat twice daily dosing. A decrease in exposure was observed with repeat dosing, likely due to induction of its own metabolism. Mean accumulation AUC Day 18/Day 1 ratios was 0.73. Following administration of 150 mg twice daily, geometric mean C
max, AUC
(0-τ) and predose concentration (Cτ) were 1478 ng/ml, 4341 ng*hr/ml and 26 ng/ml, respectively.
Administration of dabrafenib with food reduced the bioavailability (C
max and AUC decreased by 51% and 31% respectively) and delayed absorption of dabrafenib capsules when compared to the fasted state.
Distribution: Dabrafenib binds to human plasma protein and is 99.7% bound. The steady-state volume of distribution following intravenous microdose administration is 46 L.
Biotransformation: The metabolism of dabrafenib is primarily mediated by CYP2C8 and CYP3A4 to form hydroxy-dabrafenib, which is further oxidised via CYP3A4 to form carboxy-dabrafenib. Carboxy-dabrafenib can be decarboxylated via a non-enzymatic process to form desmethyl-dabrafenib. Carboxy-dabrafenib is excreted in bile and urine. Desmethyl-dabrafenib may also be formed in the gut and reabsorbed. Desmethyl-dabrafenib is metabolised by CYP3A4 to oxidative metabolites. Hydroxy-dabrafenib terminal half-life parallels that of parent with a half-life of 10 hrs while the carboxy- and desmethyl-metabolites exhibited longer half-lives (21-22 hours). Mean metabolite to parent AUC ratios following repeat-dose administration were 0.9, 11 and 0.7 for hydroxy-, carboxy-, and desmethyl-dabrafenib, respectively. Based on exposure, relative potency, and pharmacokinetic properties, both hydroxy- and desmethyl-dabrafenib are likely to contribute to the clinical activity of dabrafenib while the activity of carboxy-dabrafenib is not likely to be significant.
In vitro evaluation of drug-drug interaction potential: Dabrafenib is a substrate of human P-glycoprotein (Pgp) and human BCRP
in vitro. However, these transporters have minimal impact on dabrafenib oral bioavailability and elimination and the risk for clinically relevant drug-drug interactions with inhibitors of Pgp or BCRP is low. Neither dabrafenib nor its 3 main metabolites were demonstrated to be inhibitors of Pgp
in vitro.
Although dabrafenib and its metabolites, hydroxy-dabrafenib, carboxy-dabrafenib and desmethyl-dabrafenib, were inhibitors of human organic anion transporter (OAT) 1 and OAT3
in vitro, and dabrafenib and its desmethyl metabolite were found to be inhibitors of organic cation transporter 2 (OCT2)
in vitro, the risk of a drug-drug interaction at these transporters is minimal based on clinical exposure of dabrafenib and its metabolites.
Elimination: Terminal half-life of dabrafenib following an intravenous single microdose is 2.6 hours. Dabrafenib terminal half-life after a single oral dose is 8 hours due to absorption-limited elimination after oral administration (flip-flop pharmacokinetics). IV plasma clearance is 12 l/hr.
After an oral dose, the major route of elimination of dabrafenib is metabolism, mediated via CYP3A4 and CYP2C8. Dabrafenib related material is excreted primarily in faeces, with 71% of an oral dose recovered in faeces; 23% of the dose was recovered in urine in the form of metabolites only.
Special patient populations: Hepatic impairment: A population pharmacokinetic analysis indicates that mildly elevated bilirubin and/or AST levels (based on National Cancer Institute [NCI] classification) do not significantly affect dabrafenib oral clearance. In addition, mild hepatic impairment as defined by bilirubin and AST did not have a significant effect on dabrafenib metabolite plasma concentrations. No data are available in patients with moderate to severe hepatic impairment. As hepatic metabolism and biliary secretion are the primary routes of elimination of dabrafenib and its metabolites, administration of dabrafenib should be undertaken with caution in patients with moderate to severe hepatic impairment (see Dosage & Administration).
Renal impairment: A population pharmacokinetic analysis suggests that mild renal impairment does not affect oral clearance of dabrafenib. Although data in moderate renal impairment are limited these data may indicate no clinically relevant effect. No data are available in subjects with severe renal impairment (see Dosage & Administration).
Elderly: Based on the population pharmacokinetic analysis, age had no significant effect on dabrafenib pharmacokinetics. Age greater than 75 years was a significant predictor of carboxy- and desmethyl-dabrafenib plasma concentrations with a 40% greater exposure in subjects ≥75 years of age, relative to subjects <75 years old.
Body weight and gender: Based on the population pharmacokinetic analysis, gender and weight were found to influence dabrafenib oral clearance; weight also impacted oral volume of distribution and distributional clearance. These pharmacokinetic differences were not considered clinically relevant.
Race: The population pharmacokinetic analysis showed no significant differences in the pharmacokinetics of dabrafenib between Asian and Caucasian patients. There are insufficient data to evaluate the potential effect of other races on dabrafenib pharmacokinetics.
Paediatric population: No studies have been conducted to investigate the pharmacokinetics of dabrafenib in paediatric patients.
Toxicology: Preclinical safety data: Carcinogenicity studies with dabrafenib have not been conducted. Dabrafenib was not mutagenic or clastogenic using
in vitro tests in bacteria and cultured mammalian cells, and an
in vivo rodent micronucleus assay.
In combined female fertility, early embryonic and embryo-foetal development studies in rats numbers of ovarian corpora lutea were reduced in pregnant females at 300 mg/kg/day (approximately 3 times human clinical exposure based on AUC), but there were no effects on oestrous cycle, mating or fertility indices. Developmental toxicity including embryo-lethality and ventricular septal defects and variation in thymic shape were seen at 300 mg/kg/day, and delayed skeletal development and reduced foetal body weight at ≥20 mg/kg/day (≥0.5 times human clinical exposure based on AUC).
Male fertility studies with dabrafenib have not been conducted. However, in repeat dose studies, testicular degeneration/depletion was seen in rats and dogs (≥0.2 times the human clinical exposure based on AUC). Testicular changes in rat and dog were still present following a 4-week recovery period (see Use in Pregnancy & Lactation).
Cardiovascular effects, including coronary arterial degeneration/necrosis and/or haemorrhage, cardiac atrioventricular valve hypertrophy/haemorrhage and atrial fibrovascular proliferation were seen in dogs (≥2 times clinical exposure based on AUC). Focal arterial/perivascular inflammation in various tissues was observed in mice and an increased incidence of hepatic arterial degeneration and spontaneous cardiomyocyte degeneration with inflammation (spontaneous cardiomyopathy) was observed in rats (≥0.5 and 0.6 times clinical exposure for rats and mice respectively). Hepatic effects, including hepatocellular necrosis and inflammation, were observed in mice (≥0.6 times clinical exposure). Bronchoalveolar inflammation of the lungs was observed in several dogs at ≥20 mg/kg/day (≥9 times human clinical exposure based on AUC) and was associated with shallow and/or laboured breathing.
Reversible haematological effects have been observed in dogs and rats given dabrafenib. In studies of up to 13 weeks, decreases in reticulocyte counts and/or red cell mass were observed in dogs and rats (≥10 and 1.4 times clinical exposure, respectively).
In juvenile toxicity studies in rats, effects on growth (shorter long bone length), renal toxicity (tubular deposits, increased incidence of cortical cysts and tubular basophilia and reversible increases in urea and/or creatinine concentrations) and testicular toxicity (degeneration and tubular dilation) were observed (≥0.2 times adult human clinical exposure based on AUC).
Dabrafenib was phototoxic in an
in vitro mouse fibroblast 3T3 Neutral Red Uptake (NRU) assay and
in vivo at doses ≥100 mg/kg (>44 times clinical exposure based on C
max) in an oral phototoxicity study in hairless mice.
Combination with trametinib: In a study in dogs in which trametinib and dabrafenib were given in combination for 4 weeks, signs of gastrointestinal toxicity and decreased lymphoid cellularity of the thymus were observed at lower exposures than in dogs given trametinib alone. Otherwise, similar toxicities were observed as in comparable monotherapy studies.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image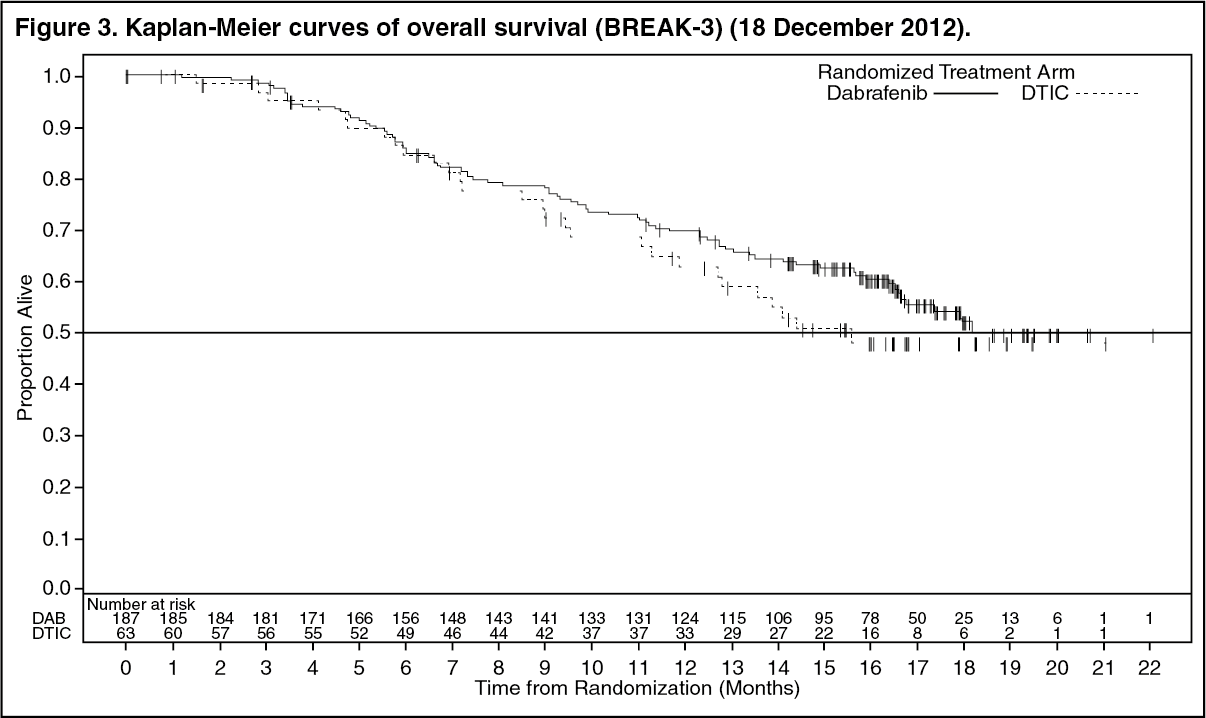 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out
