Pharmacotherapeutic group: Immunosuppressants, interleukin inhibitors.
ATC code: L04AC05.
Pharmacology: Pharmacodynamics: Mechanism of action: Ustekinumab is a fully human IgG1κ monoclonal antibody that binds with specificity to the shared p40 protein subunit of human cytokines interleukin (IL)-12 and IL-23. Ustekinumab inhibits the bioactivity of human IL-12 and IL-23 by preventing p40 from binding to the IL-12Rβ1 receptor protein expressed on the surface of immune cells. Ustekinumab cannot bind to IL-12 or IL-23 that is already bound to IL-12Rβ1 cell surface receptors. Thus, ustekinumab is not likely to contribute to complement- or antibody-mediated cytotoxicity of cells with IL-12 and/or IL-23 receptors. IL-12 and IL-23 are heterodimeric cytokines secreted by activated antigen presenting cells, such as macrophages and dendritic cells, and both cytokines participate in immune functions; IL-12 stimulates natural killer (NK) cells and drives the differentiation of CD4+ T cells toward the T helper 1 (Th1) phenotype, IL-23 induces the T helper 17 (Th17) pathway. However, abnormal regulation of IL 12 and IL 23 has been associated with immune mediated diseases, such as psoriasis, psoriatic arthritis, Crohn's disease and ulcerative colitis.
By binding the shared p40 subunit of IL-12 and IL-23, ustekinumab may exert its clinical effects in psoriasis, psoriatic arthritis, Crohn's disease and ulcerative colitis through interruption of the Th1 and Th17 cytokine pathways, which are central to the pathology of these diseases.
In patients with Crohn's disease, treatment with ustekinumab resulted in a decrease in inflammatory markers including C-Reactive Protein (CRP) and fecal calprotectin during the induction phase, which were then maintained throughout the maintenance phase. CRP was assessed during the study extension and the reductions observed during maintenance were generally sustained through week 252.
In patients with ulcerative colitis, treatment with ustekinumab resulted in a decrease in inflammatory markers including CRP and fecal calprotectin during the induction phase, which was maintained throughout the maintenance phase and study extension through week 92.
Immunisation: During the long term extension of Psoriasis Study 2 (PHOENIX 2), adult patients treated with STELARA for at least 3.5 years mounted similar antibody responses to both pneumococcal polysaccharide and tetanus vaccines as a non-systemically treated psoriasis control group. Similar proportions of adult patients developed protective levels of anti-pneumococcal and anti-tetanus antibodies and antibody titers were similar among STELARA-treated and control patients.
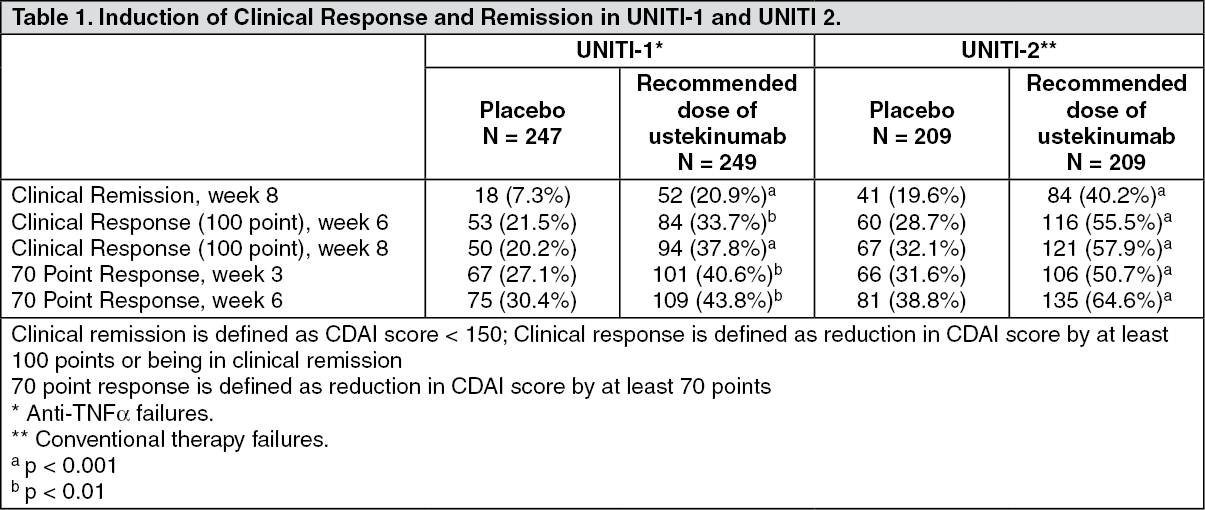
Clinical efficacy: Crohn's Disease: The safety and efficacy of ustekinumab was assessed in three randomized, double-blind, placebo-controlled, multicenter studies in adult patients with moderately to severely active Crohn's disease (Crohn's Disease Activity Index [CDAI] score of ≥ 220 and ≤ 450). The clinical development program consisted of two 8-week intravenous induction studies (UNITI-1 and UNITI-2) followed by a 44 week subcutaneous randomized withdrawal maintenance study (IM-UNITI) representing 52 weeks of therapy.
The induction studies included 1409 (UNITI-1, n = 769; UNITI-2 n = 640) patients. The primary endpoint for both induction studies was the proportion of subjects in clinical response (defined as a reduction in CDAI score of ≥ 100 points) at week 6. Efficacy data were collected and analyzed through week 8 for both studies. Concomitant doses of oral corticosteroids, immunomodulators, aminosalicylates and antibiotics were permitted and 75% of patients continued to receive at least one of these medications. In both studies, patients were randomised to receive a single intravenous administration of either the recommended tiered dose of approximately 6 mg/kg (see Table 10 in Dosage & Administration), a fixed dose of 130 mg ustekinumab, or placebo at week 0.
Patients in UNITI-1 had failed or were intolerant to prior anti-TNFα therapy. Approximately 48% of the patients had failed 1 prior anti-TNFα therapy and 52% had failed 2 or 3 prior anti-TNFα therapies. In this study, 29.1% of the patients had an inadequate initial response (primary non-responders), 69.4% responded but lost response (secondary non-responders), and 36.4% were intolerant to anti-TNFα therapies.
Patients in UNITI-2 had failed at least one conventional therapy, including corticosteroids or immunomodulators, and were either anti-TNF-α naïve (68.6%) or had previously received but not failed anti-TNFα therapy (31.4%).
In both UNITI-1 and UNITI-2, a significantly greater proportion of patients were in clinical response and remission in the ustekinumab treated group compared to placebo (Table 1). Clinical response and remission were significant as early as week 3 in ustekinumab treated patients and continued to improve through week 8. In these induction studies, efficacy was higher and better sustained in the tiered dose group compared to the 130 mg dose group, and tiered dosing is therefore the recommended intravenous induction dose. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The maintenance study (IM-UNITI), evaluated 388 patients who achieved 100 point clinical response at week 8 of induction with ustekinumab in studies UNITI-1 and UNITI-2. Patients were randomized to receive a subcutaneous maintenance regimen of either 90 mg ustekinumab every 8 weeks, 90 mg ustekinumab every 12 weeks or placebo for 44 weeks (for recommended maintenance posology, see Solution for injection (SC) under Dosage & Administration).
Significantly higher proportions of patients maintained clinical remission and response in the ustekinumab treated groups compared to the placebo group at week 44 (see Table 2).
Click on icon to see table/diagram/image
In IM-UNITI, 29 of 129 patients did not maintain response to ustekinumab when treated every 12 weeks and were allowed to dose adjust to receive ustekinumab every 8 weeks. Loss of response was defined as a CDAI score ≥ 220 points and a ≥ 100 point increase from the CDAI score at baseline. In these patients, clinical remission was achieved in 41.4% of patients 16 weeks after dose adjustment.
Patients who were not in clinical response to ustekinumab induction at week 8 of the UNITI-1 and UNITI-2 induction studies (476 patients) entered into the non-randomized portion of the maintenance study (IM-UNITI) and received a 90 mg subcutaneous injection of ustekinumab at that time. Eight weeks later, 50.5% of the patients achieved clinical response and continued to receive maintenance dosing every 8 weeks; among these patients with continued maintenance dosing, a majority maintained response (68.1%) and achieved remission (50.2%) at week 44, at proportions that were similar to the patients who initially responded to ustekinumab induction.
Of 131 patients who responded to ustekinumab induction, and were randomized to the placebo group at the start of the maintenance study, 51 subsequently lost response and received 90 mg ustekinumab subcutaneously every 8 weeks. The majority of patients who lost response and resumed ustekinumab did so within 24 weeks of the induction infusion. Of these 51 patients, 70.6% achieved clinical response and 39.2% percent achieved clinical remission 16 weeks after receiving the first subcutaneous dose of ustekinumab.
In IM-UNITI, patients who completed the study through week 44 were eligible to continue treatment in a study extension. Among the 718 patients who entered and were treated in the study extension, clinical remission and response were generally maintained through week 252 for both patients who failed TNF-therapies and those who failed conventional therapies.
No new safety concerns were identified in this study extension with up to 5 years of treatment in patients with Crohn's Disease.
Endoscopy: Endoscopic appearance of the mucosa was evaluated in 252 patients with eligible baseline endoscopic disease activity in a substudy. The primary endpoint was change from baseline in Simplified Endoscopic Disease Severity Score for Crohn's Disease (SES-CD), a composite score across 5 ileo-colonic segments of presence/size of ulcers, proportion of mucosal surface covered by ulcers, proportion of mucosal surface affected by any other lesions and presence/type of narrowing/strictures. At week 8, after a single intravenous induction dose, the change in SES-CD score was greater in the ustekinumab group (n = 155, mean change = -2.8) than in the placebo group (n = 97, mean change = -0.7, p = 0.012).
Fistula Response: In a subgroup of patients with draining fistulas at baseline (8.8%; n = 26), 12/15 (80%) of ustekinumab-treated patients achieved a fistula response over 44 weeks (defined as ≥ 50% reduction from baseline of the induction study in the number of draining fistulas) compared to 5/11 (45.5%) exposed to placebo.
Health-related quality of life: Health-related quality of life was assessed by Inflammatory Bowel Disease Questionnaire (IBDQ) and SF-36 questionnaires. At week 8, patients receiving ustekinumab showed statistically significantly greater and clinically meaningful improvements on IBDQ total score and SF-36 Mental Component Summary Score in both UNITI-1 and UNITI-2, and SF-36 Physical Component Summary Score in UNITI-2, when compared to placebo. These improvements were generally better maintained in ustekinumab-treated patients in the IM-UNITI study through week 44 when compared to placebo. Improvement in health-related quality of life was generally maintained during the extension through week 252.
Ulcerative colitis: The safety and efficacy of ustekinumab was assessed in two randomized, double-blind, placebo-controlled, multicenter studies in adult patients with moderately to severely active ulcerative colitis (Mayo score 6 to 12; Endoscopy subscore ≥ 2). The clinical development program consisted of one intravenous induction study (referred to as UNIFI-I) with treatment of up to 16 weeks followed by a 44 week subcutaneous randomized withdrawal maintenance study (referred to as UNIFI-M) representing at least 52 weeks of therapy.
Efficacy results presented for UNIFI-I and UNIFI-M were based on central review of endoscopies.
UNIFI-I included 961 patients. The primary endpoint for the induction study was the proportion of subjects in clinical remission at week 8. Patients were randomised to receive a single intravenous administration of either the recommended tiered dose of approximately 6 mg/kg (see Table 10 in Dosage & Administration), a fixed dose of 130 mg ustekinumab, or placebo at week 0.
Concomitant doses of oral corticosteroids, immunomodulators, and aminosalicylates were permitted and 90% of patients continued to receive at least one of these medications. Enrolled patients had to have failed conventional therapy (corticosteroids or immunomodulators) or at least one biologic (a TNFα antagonist and/or vedolizumab). 49% of patients had failed conventional therapy, but not a biologic (of which 94% where biological-naïve). 51% of patients had failed or were intolerant to a biologic. Approximately 50% of the patients had failed at least 1 prior anti-TNFα therapy (of which 48% were primary non-responders) and 17% had failed at least 1 anti-TNFα therapy and vedolizumab.
In UNIFI-I a significantly greater proportion of patients were in clinical remission in the ustekinumab treated group compared to placebo at week 8 (Table 3). As early as Week 2, the earliest scheduled study visit, and at each visit thereafter, a higher proportion of ustekinumab patients had no rectal bleeding or achieved normal stool frequency as compared with placebo patients. Significant differences in partial Mayo score and symptomatic remission were observed between ustekinumab and placebo as early as Week 2.
Efficacy was higher in the tiered dose group (6 mg/kg) compared to the 130 mg dose group in select endpoints, and tiered dosing is therefore the recommended intravenous induction dose. (See Table 3.)
Click on icon to see table/diagram/image
UNIFI-M, evaluated 523 patients who achieved clinical response with single IV administration of ustekinumab in UNIFI-I. Patients were randomized to receive a subcutaneous maintenance regimen of either 90 mg ustekinumab every 8 weeks, 90 mg ustekinumab every 12 weeks or placebo for 44 weeks (for recommended maintenance posology, see Solution for injection (SC) under Dosage & Administration).
Significantly greater proportions of patients were in clinical remission in both ustekinumab treated groups compared to the placebo group at week 44 (see Table 4).
Click on icon to see table/diagram/image
The beneficial effect of ustekinumab on clinical response, mucosal healing and clinical remission was observed in induction and in maintenance both in patients who failed conventional therapy but not a biologic therapy, as well as in those who had failed at least one prior TNFα antagonist therapy including in patients with a primary non-response to TNFα antagonist therapy. A beneficial effect was also observed in induction in patients who failed at least one prior TNFα antagonist therapy and vedolizumab, however the number of patients in this subgroup was too small to draw definitive conclusions about the beneficial effect in this group during maintenance.
Week 16 Responders to Ustekinumab Induction: Ustekinumab treated patients who were not in response at week 8 of UNIFI-I received an administration of 90 mg SC ustekinumab at week 8 (36% of patients). Of those patients, 9% of patients who were initially randomized to the recommended induction dose achieved clinical remission and 58% achieved clinical response at Week 16.
Patients who were not in clinical response to ustekinumab induction at week 8 of the UNIFI-I study but were in response at week 16 (157 patients) entered into the non-randomized portion of UNIFI-M and continued to receive maintenance dosing every 8 weeks; among these patients, a majority (62%) maintained response and 30% achieved remission at week 44.
Study Extension: In UNIFI, patients who completed the study through week 44 were eligible to continue treatment in a study extension. Among the 588 patients who entered and were treated in the study extension, symptomatic remission was generally maintained through week 92 for patients who failed conventional therapy (but not a biologic therapy) and those who failed biologic therapy, including those who failed both anti-TNF and vedolizumab.
No new safety concerns were identified in this study extension with up to 2 years of treatment in patients with ulcerative colitis.
Endoscopic Normalization: Endoscopic normalization was defined as a Mayo endoscopic subscore of 0 and was observed as early as week 8 of UNIFI-I. At week 44 of UNIFI-M, it was achieved in 24% and 29% of patients treated with ustekinumab every 12 or 8 weeks, respectively, as compared to 18% of patients in the placebo group.
Histologic & Histo-Endoscopic Mucosal Healing: Histologic healing (defined as neutrophil infiltration in < 5% of crypts, no crypt destruction, and no erosions, ulcerations, or granulation tissue) was assessed at week 8 of UNIFI-I and Week 44 of UNIFI-M. At week 8, after a single intravenous induction dose, significantly greater proportions of patients in the recommended dose group achieved histologic healing (36%) compared with patients in the placebo group (22%). At Week 44 maintenance of this effect was observed with significantly more patients in histologic healing in the every 12 week (54%) and every 8 week (59%) ustekinumab groups as compared to placebo (33%).
A combined endpoint of histo-endoscopic mucosal healing defined as subjects having both mucosal healing and histologic healing was evaluated at week 8 of UNIFI-I and week 44 of UNIFI-M. Patients receiving ustekinumab at the recommended dose showed significant improvements on the histo-endoscopic mucosal healing endpoint at week 8 in the ustekinumab group (18%) as compared to the placebo group (9%). At week 44, maintenance of this effect was observed with significantly more patients in histo-endoscopic mucosal healing in the every 12 week (39%) and every 8 week (46%) ustekinumab groups as compared to placebo (24%).
Health-related quality of life: Health-related quality of life was assessed by Inflammatory Bowel Disease Questionnaire (IBDQ), SF-36 and EuroQoL-5D (EQ-5D) questionnaires.
At week 8 of UNIFI-I, patients receiving ustekinumab showed significantly greater and clinically meaningful improvements on IBDQ total score, EQ-5D and EQ-5D VAS, and SF-36 Mental Component Summary Score and SF-36 Physical Component Summary Score when compared to placebo. These improvements were maintained in ustekinumab-treated patients in UNIFI-M through week 44. Improvement in health-related quality of life as measured by IBDQ and SF-36 was generally maintained during the extension through week 92.
Patients receiving ustekinumab experienced significantly more improvements in work productivity as assessed by greater reductions in overall work impairment and in activity impairment as assessed by the WPAI-GH questionnaire than patients receiving placebo.
Hospitalizations and ulcerative colitis (UC) related surgeries: Through week 8 of UNIFI-I, the proportions of subjects with UC disease related hospitalizations were significantly lower for subjects in the ustekinumab recommended dose group (1.6%, 5/322) compared with subjects in the placebo group (4.4%, 14/319) and no subjects underwent UC disease related surgeries in subjects receiving ustekinumab at the recommended induction dose compared to 0.6% (2/319) subjects in the placebo group.
Through week 44 of UNIFI-M, a significantly lower number of UC-related hospitalizations was observed in subjects in the combined ustekinumab group (2.0%, 7/348) as compared with subjects in the placebo group (5.7%, 10/175). A numerically lower number of subjects in the ustekinumab group (0.6%, 2/348) underwent UC disease related surgeries compared with subjects in the placebo group (1.7%, 3/175) through week 44.
Solution for injection (SC): Plaque psoriasis (Adults): The safety and efficacy of ustekinumab was assessed in 1,996 patients in two randomised, double-blind, placebo-controlled studies in patients with moderate to severe plaque psoriasis and who were candidates for phototherapy or systemic therapy. In addition, a randomised, blinded assessor, active-controlled study compared ustekinumab and etanercept in patients with moderate to severe plaque psoriasis who had had an inadequate response to, intolerance to, or contraindication to ciclosporin, MTX, or PUVA.
Psoriasis Study 1 (PHOENIX 1) evaluated 766 patients. 53% of these patients were either non-responsive, intolerant, or had a contraindication to other systemic therapy. Patients randomised to ustekinumab received 45 mg or 90 mg doses at Weeks 0 and 4 and followed by the same dose every 12 weeks. Patients randomised to receive placebo at Weeks 0 and 4 crossed over to receive ustekinumab (either 45 mg or 90 mg) at Weeks 12 and 16 followed by dosing every 12 weeks. Patients originally randomised to ustekinumab who achieved Psoriasis Area and Severity Index 75 response (PASI improvement of at least 75% relative to baseline) at both Weeks 28 and 40 were re-randomised to receive ustekinumab every 12 weeks or to placebo (i.e., withdrawal of therapy). Patients who were re-randomised to placebo at week 40 reinitiated ustekinumab at their original dosing regimen when they experienced at least a 50% loss of their PASI improvement obtained at week 40. All patients were followed for up to 76 weeks following first administration of study treatment.
Psoriasis Study 2 (PHOENIX 2) evaluated 1,230 patients. 61% of these patients were either non-responsive, intolerant, or had a contraindication to other systemic therapy. Patients randomised to ustekinumab received 45 mg or 90 mg doses at Weeks 0 and 4 followed by an additional dose at 16 weeks. Patients randomised to receive placebo at Weeks 0 and 4 crossed over to receive ustekinumab (either 45 mg or 90 mg) at Weeks 12 and 16. All patients were followed for up to 52 weeks following first administration of study treatment.
Psoriasis Study 3 (ACCEPT) evaluated 903 patients with moderate to severe psoriasis who inadequately responded to, were intolerant to, or had a contraindication to other systemic therapy and compared the efficacy of ustekinumab to etanercept and evaluated the safety of ustekinumab and etanercept. During the 12-week active-controlled portion of the study, patients were randomised to receive etanercept (50 mg twice a week), ustekinumab 45 mg at Weeks 0 and 4, or ustekinumab 90 mg at Weeks 0 and 4.
Baseline disease characteristics were generally consistent across all treatment groups in Psoriasis Studies 1 and 2 with a median baseline PASI score from 17 to 18, median baseline Body Surface Area (BSA) ≥ 20, and median Dermatology Life Quality Index (DLQI) range from 10 to 12. Approximately one third (Psoriasis Study 1) and one quarter (Psoriasis Study 2) of subjects had Psoriatic Arthritis (PsA). Similar disease severity was also seen in Psoriasis Study 3.
The primary endpoint in these studies was the proportion of patients who achieved PASI 75 response from baseline at week 12 (see Tables 5 and 6).
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In Psoriasis Study 1 maintenance of PASI 75 was significantly superior with continuous treatment compared with treatment withdrawal (p < 0.001). Similar results were seen with each dose of ustekinumab. At 1 year (week 52), 89% of patients re-randomised to maintenance treatment were PASI 75 responders compared with 63% of patients re-randomised to placebo (treatment withdrawal) (p < 0.001). At 18 months (week 76), 84% of patients re-randomised to maintenance treatment were PASI 75 responders compared with 19% of patients re-randomised to placebo (treatment withdrawal). At 3 years (week 148), 82% of patients re-randomised to maintenance treatment were PASI 75 responders. At 5 years (week 244), 80% of patients re-randomised to maintenance treatment were PASI 75 responders.
In patients re-randomised to placebo, and who reinitiated their original ustekinumab treatment regimen after loss of ≥ 50% of PASI improvement 85% regained PASI 75 response within 12 weeks after re-initiating therapy.
In Psoriasis Study 1, at week 2 and week 12, significantly greater improvements from baseline were demonstrated in the DLQI in each ustekinumab treatment group compared with placebo. The improvement was sustained through week 28. Similarly, significant improvements were seen in Psoriasis Study 2 at week 4 and 12, which were sustained through week 24. In Psoriasis Study 1, improvements in nail psoriasis (Nail Psoriasis Severity Index), in the physical and mental component summary scores of the SF-36 and in the Itch Visual Analogue Scale (VAS) were also significant in each ustekinumab treatment group compared with placebo. In Psoriasis Study 2, the Hospital Anxiety and Depression Scale (HADS) and Work Limitations Questionnaire (WLQ) were also significantly improved in each ustekinumab treatment group compared with placebo.
Psoriatic arthritis (PsA) (Adults): Ustekinumab has been shown to improve signs and symptoms, physical function and health-related quality of life, and reduce the rate of progression of peripheral joint damage in adult patients with active PsA.
The safety and efficacy of ustekinumab was assessed in 927 patients in two randomised, double-blind, placebo-controlled studies in patients with active PsA (≥ 5 swollen joints and ≥ 5 tender joints) despite non-steroidal anti-inflammatory (NSAID) or disease modifying antirheumatic (DMARD) therapy. Patients in these studies had a diagnosis of PsA for at least 6 months. Patients with each subtype of PsA were enrolled, including polyarticular arthritis with no evidence of rheumatoid nodules (39%), spondylitis with peripheral arthritis (28%), asymmetric peripheral arthritis (21%), distal interphalangeal involvement (12%) and arthritis mutilans (0.5%). Over 70% and 40% of the patients in both studies had enthesitis and dactylitis at baseline, respectively. Patients were randomised to receive treatment with ustekinumab 45 mg, 90 mg, or placebo subcutaneously at Weeks 0 and 4 followed by every 12 weeks (q12w) dosing. Approximately 50% of patients continued on stable doses of MTX (≤ 25 mg/week).
In PsA Study 1 (PSUMMIT I) and PsA Study 2 (PSUMMIT II), 80% and 86% of the patients, respectively, had been previously treated with DMARDs. In Study 1 previous treatment with anti-tumour necrosis factor (TNF)α agent was not allowed. In Study 2, the majority of patients (58%, n = 180) had been previously treated with one or more anti-TNFα agent(s), of whom over 70% had discontinued their anti-TNFα treatment for lack of efficacy or intolerance at any time.
Signs and symptoms: Treatment with ustekinumab resulted in significant improvements in the measures of disease activity compared to placebo at week 24. The primary endpoint was the percentage of patients who achieved American College of Rheumatology (ACR) 20 response at week 24. The key efficacy results are shown in Table 7 as follows. (See Table 7.)
Click on icon to see table/diagram/image
ACR 20, 50 and 70 responses continued to improve or were maintained through week 52 (PsA Study 1 and 2) and week 100 (PsA Study 1). In PsA Study 1, ACR 20 responses at week 100 were achieved by 57% and 64%, for 45 mg and 90 mg, respectively. In PsA Study 2, ACR 20 responses at week 52 were achieved by 47% and 48%, for 45 mg and 90 mg, respectively.
The proportion of patients achieving a modified PsA response criteria (PsARC) response was also significantly greater in the ustekinumab groups compared to placebo at week 24. PsARC responses were maintained through weeks 52 and 100. A higher proportion of patients treated with ustekinumab who had spondylitis with peripheral arthritis as their primary presentation, demonstrated 50 and 70 percent improvement in Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) scores compared with placebo at week 24.
Responses observed in the ustekinumab treated groups were similar in patients receiving and not receiving concomitant MTX, and were maintained through weeks 52 and 100. Patients previously treated with anti-TNFα agents who received ustekinumab achieved a greater response at week 24 than patients receiving placebo (ACR 20 response at week 24 for 45 mg and 90 mg was 37% and 34%, respectively, compared with placebo 15%; p < 0.05), and responses were maintained through week 52.
For patients with enthesitis and/or dactylitis at baseline, in PsA Study 1 significant improvement in enthesitis and dactylitis score was observed in the ustekinumab groups compared with placebo at week 24. In PsA Study 2 significant improvement in enthesitis score and numerical improvement (not statistically significant) in dactylitis score was observed in the ustekinumab 90 mg group compared with placebo at week 24. Improvements in enthesitis score and dactylitis score were maintained through weeks 52 and 100.
Radiographic Response: Structural damage in both hands and feet was expressed as change in total van der Heijde-Sharp score (vdH-S score), modified for PsA by addition of hand distal interphalangeal joints, compared to baseline. A pre-specified integrated analysis combining data from 927 subjects in both PsA Study 1 and 2 was performed. Ustekinumab demonstrated a statistically significant decrease in the rate of progression of structural damage compared to placebo, as measured by change from baseline to week 24 in the total modified vdH-S score (mean ± SD score was 0.97 ± 3.85 in the placebo group compared with 0.40 ± 2.11 and 0.39 ± 2.40 in the ustekinumab 45 mg (p < 0.05) and 90 mg (p < 0.001) groups, respectively). This effect was driven by PsA Study 1. The effect is considered demonstrated irrespective of concomitant MTX use, and was maintained through Weeks 52 (integrated analysis) and 100 (PsA Study 1).
Physical function and health-related quality of life: Ustekinumab-treated patients showed significant improvement in physical function as assessed by the Disability Index of the Health Assessment Questionnaire (HAQ-DI) at week 24. The proportion of patients achieving a clinically meaningful ≥ 0.3 improvement in HAQ-DI score from baseline was also significantly greater in the ustekinumab groups when compared with placebo. Improvement in HAQ-DI score from baseline was maintained through Weeks 52 and 100.
There was significant improvement in DLQI scores in the ustekinumab groups as compared with placebo at week 24, which was maintained through weeks 52 and 100. In PsA Study 2 there was a significant improvement in Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-F) scores in the ustekinumab groups when compared with placebo at week 24. The proportion of patients achieving a clinically significant improvement in fatigue (4 points in FACIT-F) was also significantly greater in the ustekinumab groups compared with placebo. Improvements in FACIT scores were maintained through week 52.
Paediatric population: Paediatric plaque psoriasis: Ustekinumab has been shown to improve signs and symptoms, and health-related quality of life in paediatric patients 6 years and older with plaque psoriasis.
Adolescent patients (12-17 years): The efficacy of ustekinumab was studied in 110 paediatric patients aged 12 to 17 years with moderate to severe plaque psoriasis in a multicenter, phase 3, randomised, double-blind, placebo-controlled study (CADMUS). Patients were randomised to receive either placebo (n = 37), or the recommended dose of ustekinumab (see Dosage & Administration; n = 36) or half of the recommended dose of ustekinumab (n = 37) by subcutaneous injection at Weeks 0 and 4 followed by every 12 week (q12w) dosing. At week 12, placebo-treated patients crossed over to receive ustekinumab.
Patients with PASI ≥ 12, PGA ≥ 3 and BSA involvement of at least 10%, who were candidates for systemic therapy or phototherapy, were eligible for the study. Approximately 60% of the patients had prior exposure to conventional systemic therapy or phototherapy. Approximately 11% of the patients had prior exposure to biologics.
The primary endpoint was the proportion of patients who achieved a PGA score of cleared (0) or minimal (1) at week 12. Secondary endpoints included PASI 75, PASI 90, change from baseline in Children's Dermatology Life Quality Index (CDLQI), change from baseline in the total scale score of PedsQL (Paediatric Quality of Life Inventory) at week 12. At week 12, subjects treated with ustekinumab showed significantly greater improvement in their psoriasis and health-related quality of life compared with placebo (Table 8).
All patients were followed for efficacy for up to 52 weeks following first administration of study agent. The proportion of patients with a PGA score of cleared (0) or minimal (1) and the proportion achieving PASI 75 showed separation between the ustekinumab treated group and placebo at the first post-baseline visit at week 4, reaching a maximum by week 12. Improvements in PGA, PASI, CDLQI and PedsQL were maintained through week 52 (Table 8). (See Table 8.)
Click on icon to see table/diagram/image
During the placebo-controlled period through week 12, the efficacy of both the recommended and half of the recommended dose groups were generally comparable at the primary endpoint (69.4% and 67.6% respectively) although there was evidence of a dose response for higher level efficacy criteria (e.g. PGA of cleared (0), PASI 90). Beyond week 12, efficacy was generally higher and better sustained in the recommended dose group compared with half of the recommended dosage group in which a modest loss of efficacy was more frequently observed toward the end of each 12 week dosing interval. The safety profiles of the recommended dose and half of the recommended dose were comparable.
Children (6-11 years): The efficacy of ustekinumab was studied in 44 paediatric patients aged 6 to 11 years with moderate to severe plaque psoriasis in an open label, single-arm, multicenter, phase 3, study (CADMUS Jr.). Patients were treated with the recommended dose of ustekinumab (see Dosage & Administration; n = 44) by subcutaneous injection at weeks 0 and 4 followed by every 12 week (q12w) dosing.
Patients with PASI ≥ 12, PGA ≥ 3 and BSA involvement of at least 10%, who were candidates for systemic therapy or phototherapy, were eligible for the study. Approximately 43% of the patients had prior exposure to conventional systemic therapy or phototherapy. Approximately 5% of the patients had prior exposure to biologics.
The primary endpoint was the proportion of patients who achieved a PGA score of cleared (0) or minimal (1) at week 12. Secondary endpoints included PASI 75, PASI 90, and change from baseline in Children's Dermatology Life Quality Index (CDLQI) at week 12. At week 12, subjects treated with ustekinumab showed clinically meaningful improvements in their psoriasis and health-related quality of life (Table 9).
All patients were followed for efficacy for up to 52 weeks following first administration of study agent. The proportion of patients with a PGA score of cleared (0) or minimal (1) at week 12 was 77.3%. Efficacy (defined as PGA 0 or 1) was observed as early as the first post-baseline visit at week 4 and the proportion of subjects who achieved a PGA score of 0 or 1 increased through week 16 and then remained relatively stable through week 52. Improvements in PGA, PASI, and CDLQI were maintained through week 52 (Table 9). (See Table 9.)
Click on icon to see table/diagram/image
Immunogenicity: Solution for injection (SC): Antibodies to ustekinumab may develop during ustekinumab treatment and most are neutralising. The formation of anti-ustekinumab antibodies is associated with both increased clearance and reduced efficacy of ustekinumab, except in patients with Crohn's disease or ulcerative colitis where no reduced efficacy was observed. There is no apparent correlation between the presence of anti-ustekinumab antibodies and the occurrence of injection site reactions.
Concentrate for solution for infusion (IV): Antibodies to ustekinumab may develop during ustekinumab treatment and most are neutralising. The formation of anti-ustekinumab antibodies is associated with increased clearance of ustekinumab in patients with Crohn's disease or ulcerative colitis. No reduced efficacy was observed. There is no apparent correlation between the presence of anti-ustekinumab antibodies and the occurrence of injection site reactions.
Pharmacokinetics: Distribution: Median volume of distribution during the terminal phase (V
z) following a single intravenous administration to patients with psoriasis ranged from 57 to 83 mL/kg.
Biotransformation: The exact metabolic pathway for ustekinumab is unknown.
Elimination: Median systemic clearance (CL) following a single intravenous administration to patients with psoriasis ranged from 1.99 to 2.34 mL/day/kg. Median half-life (t
1/2) of ustekinumab was approximately 3 weeks in patients with ulcerative colitis, Crohn's disease, psoriasis and/or psoriatic arthritis, ranging from 15 to 32 days across all psoriasis and psoriatic arthritis studies.
Dose linearity: The systemic exposure of ustekinumab (C
max and AUC) increased in an approximately dose-proportional manner after a single intravenous administration at doses ranging from 0.09 mg/kg to 4.5 mg/kg or following a single subcutaneous administration at doses ranging from approximately 24 mg to 240 mg in patients with psoriasis.
Special populations: No pharmacokinetic data are available in patients with impaired renal or hepatic function.
In patients with Crohn's disease and ulcerative colitis, variability in ustekinumab clearance was affected by body weight, serum albumin level, sex, and antibody to ustekinumab status while body weight was the main covariate affecting the volume of distribution. Additionally in Crohn's disease, clearance was affected by C-reactive protein, TNF antagonist failure status and race (Asian versus non-Asian). The impact of these covariates was within ±20% of the typical or reference value of the respective PK parameter, thus dose adjustment is not warranted for these covariates. Concomitant use of immunomodulators did not have a significant impact on ustekinumab disposition.
Regulation of CYP450 enzymes: The effects of IL-12 or IL-23 on the regulation of CYP450 enzymes were evaluated in an
in vitro study using human hepatocytes, which showed that IL-12 and/or IL-23 at levels of 10 ng/mL did not alter human CYP450 enzyme activities (CYP1A2, 2B6, 2C9, 2C19, 2D6, or 3A4; see Interactions).
Solution for injection (SC): Absorption: The median time to reach the maximum serum concentration (t
max) was 8.5 days after a single 90 mg subcutaneous administration in healthy subjects. The median t
max values of ustekinumab following a single subcutaneous administration of either 45 mg or 90 mg in patients with psoriasis were comparable to those observed in healthy subjects.
The absolute bioavailability of ustekinumab following a single subcutaneous administration was estimated to be 57.2% in patients with psoriasis.
Elimination: In a population pharmacokinetic analysis, the apparent clearance (CL/F) and apparent volume of distribution (V/F) were 0.465 l/day and 15.7 l, respectively, in patients with psoriasis. The CL/F of ustekinumab was not impacted by gender. Population pharmacokinetic analysis showed that there was a trend towards a higher clearance of ustekinumab in patients who tested positive for antibodies to ustekinumab.
Special populations: No specific studies have been conducted in elderly patients.
The pharmacokinetics of ustekinumab were generally comparable between Asian and non-Asian patients with psoriasis and ulcerative colitis.
In the population pharmacokinetic analysis, there were no indications of an effect of tobacco or alcohol on the pharmacokinetics of ustekinumab.
Serum ustekinumab concentrations in paediatric psoriasis patients 6 to 17 years of age, treated with the recommended weight-based dose were generally comparable to those in the adult psoriasis population treated with the adult dose. Serum ustekinumab concentrations in paediatric psoriasis patients 12-17 years of age (CADMUS) treated with half of the recommended weight-based dose were generally lower than those in adults.
Single dose versus multiple doses: Serum concentration-time profiles of ustekinumab were generally predictable after single or multiple subcutaneous dose administrations. In patients with psoriasis, steady-state serum concentrations of ustekinumab were achieved by Week 28 after initial subcutaneous doses at Weeks 0 and 4 followed by doses every 12 weeks. The median steady-state trough concentration ranged from 0.21 μg/mL to 0.26 μg/mL (45 mg) and from 0.47 μg/mL to 0.49 μg/mL (90 mg). There was no apparent accumulation in serum ustekinumab concentration over time when given subcutaneously every 12 weeks.
In patients with Crohn's disease and ulcerative colitis, following an intravenous dose of ~6 mg/kg, starting at week 8, subcutaneous maintenance dosing of 90 mg ustekinumab was administered every 8 or 12 weeks. Steady state ustekinumab concentration was achieved by the start of the second maintenance dose. In patients with Crohn's disease, median steady-state trough concentrations ranged from 1.97 μg/mL to 2.24 μg/mL and from 0.61 μg/mL to 0.76 μg/mL for 90 mg ustekinumab every 8 weeks or every 12 weeks respectively. In patients with ulcerative colitis, median steady-state trough concentrations ranged from 2.69 μg/mL to 3.09 μg/mL and from 0.92 μg/mL to 1.19 μg/mL for 90 mg ustekinumab every 8 weeks or every 12 weeks. The steady-state trough ustekinumab levels resulting from 90 mg ustekinumab every 8 weeks were associated with higher clinical remission rates as compared to the steady-state trough levels following 90 mg every 12 weeks.
Impact of weight on pharmacokinetics: In a population pharmacokinetic analysis using data from patients with psoriasis, body weight was found to be the most significant covariate affecting the clearance of ustekinumab. The median CL/F in patients with weight > 100 kg was approximately 55% higher compared to patients with weight ≤ 100 kg. The median V/F in patients with weight > 100 kg was approximately 37% higher as compared to patients with weight ≤ 100 kg. The median trough serum concentrations of ustekinumab in patients with higher weight (> 100 kg) in the 90 mg group were comparable to those in patients with lower weight (≤ 100 kg) in the 45 mg group. Similar results were obtained from a confirmatory population pharmacokinetic analysis using data from patients with psoriatic arthritis.
Dosing frequency adjustment: In patients with Crohn's disease and ulcerative colitis, based on observed data and population PK analyses, randomized subjects who lost response to treatment had lower serum ustekinumab concentrations over time compared with subjects who did not lose response. In Crohn's disease, dose adjustment from 90 mg every 12 weeks to 90 mg every 8 weeks was associated with an increase in trough serum ustekinumab concentrations and an accompanying increase in efficacy. In ulcerative colitis, population PK model based simulations demonstrated that adjusting dosing from 90 mg every 12 weeks to every 8 weeks would be expected to result in a 3-fold increase in steady-state trough ustekinumab concentrations. Additionally on the basis of clinical trial data in patients with ulcerative colitis, a positive exposure-response relationship was established between trough concentrations, and clinical remission and mucosal healing.
Concentrate for solution for infusion (IV): Following the recommended intravenous induction dose, median peak serum ustekinumab concentration, observed 1 hour after the infusion, was 126.1 μg/mL in patients with Crohn's disease and 127.0 μg/mL in patients with ulcerative colitis.
Special populations: No specific studies have been conducted with intravenous ustekinumab in elderly or paediatric patients.
Toxicology: Preclinical safety data: Non-clinical data reveal no special hazard (e.g. organ toxicity) for humans based on studies of repeated-dose toxicity and developmental and reproductive toxicity, including safety pharmacology evaluations. In developmental and reproductive toxicity studies in cynomolgus monkeys, neither adverse effects on male fertility indices nor birth defects or developmental toxicity were observed. No adverse effects on female fertility indices were observed using an analogous antibody to IL-12/23 in mice.
Dose levels in animal studies were up to approximately 45-fold higher than the highest equivalent dose intended to be administered to psoriasis patients and resulted in peak serum concentrations in monkeys that were more than 100-fold higher than observed in humans.
Carcinogenicity studies were not performed with ustekinumab due to the lack of appropriate models for an antibody with no cross-reactivity to rodent IL-12/23 p40.




 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image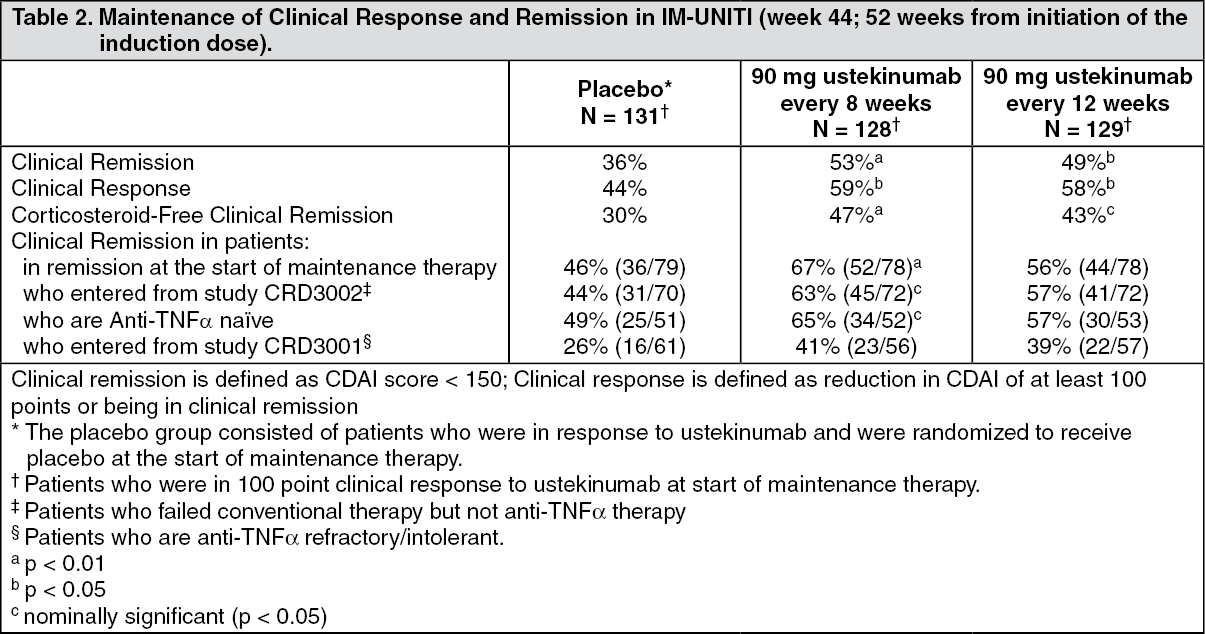 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image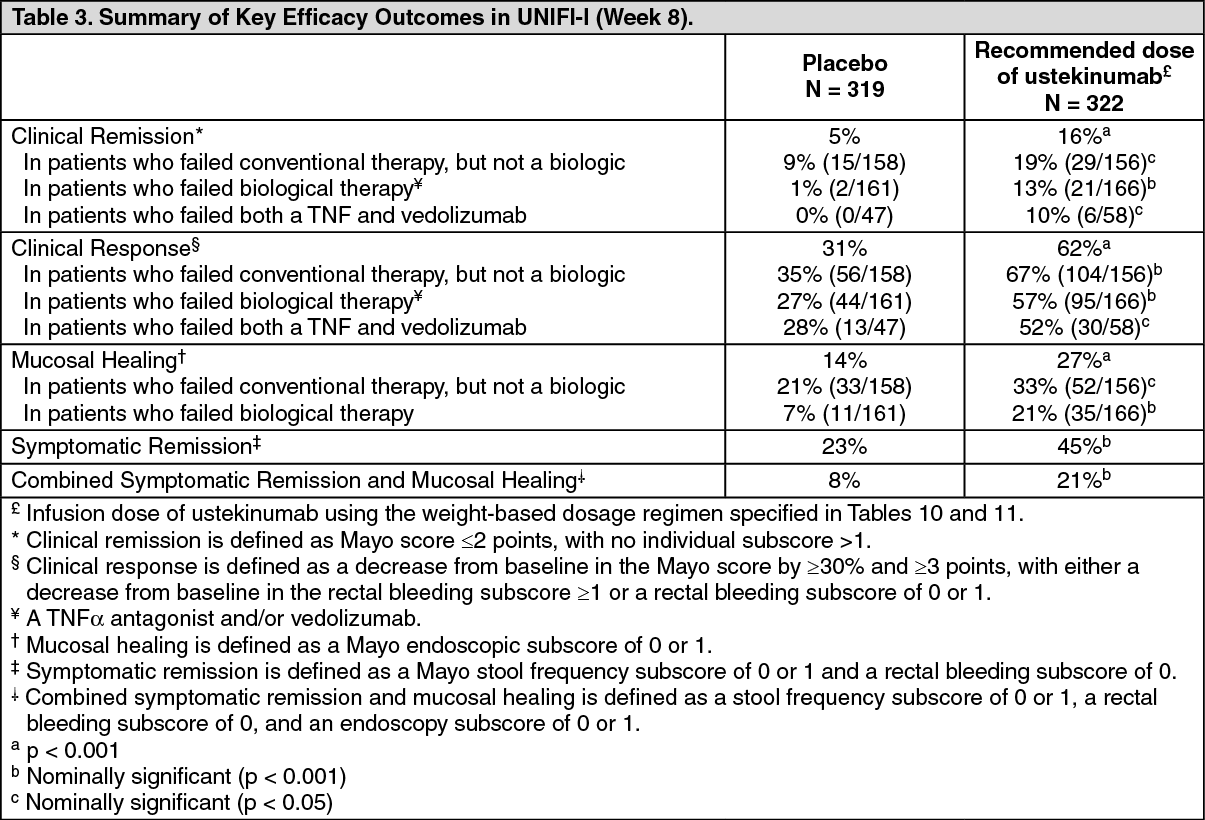 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image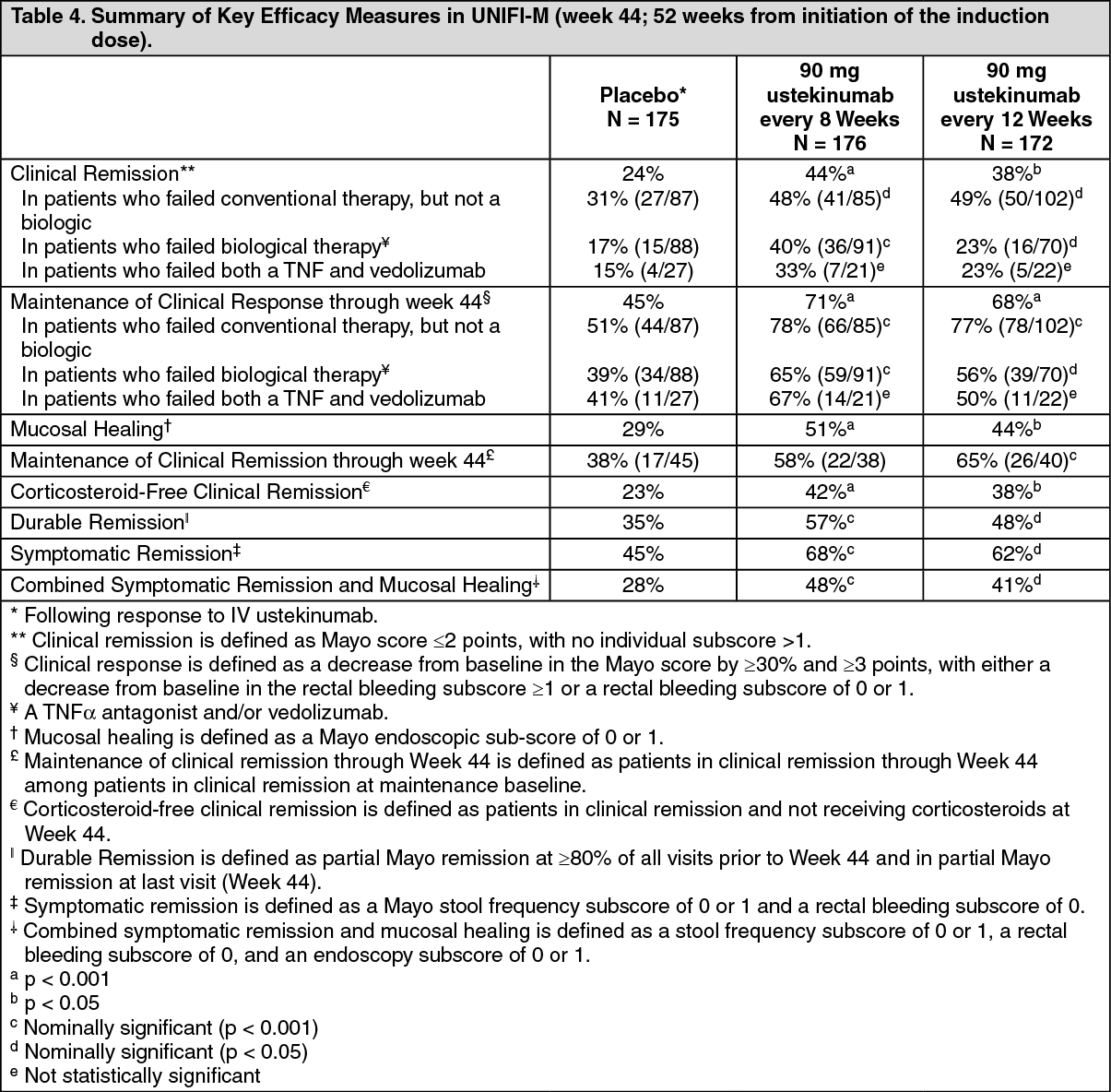 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image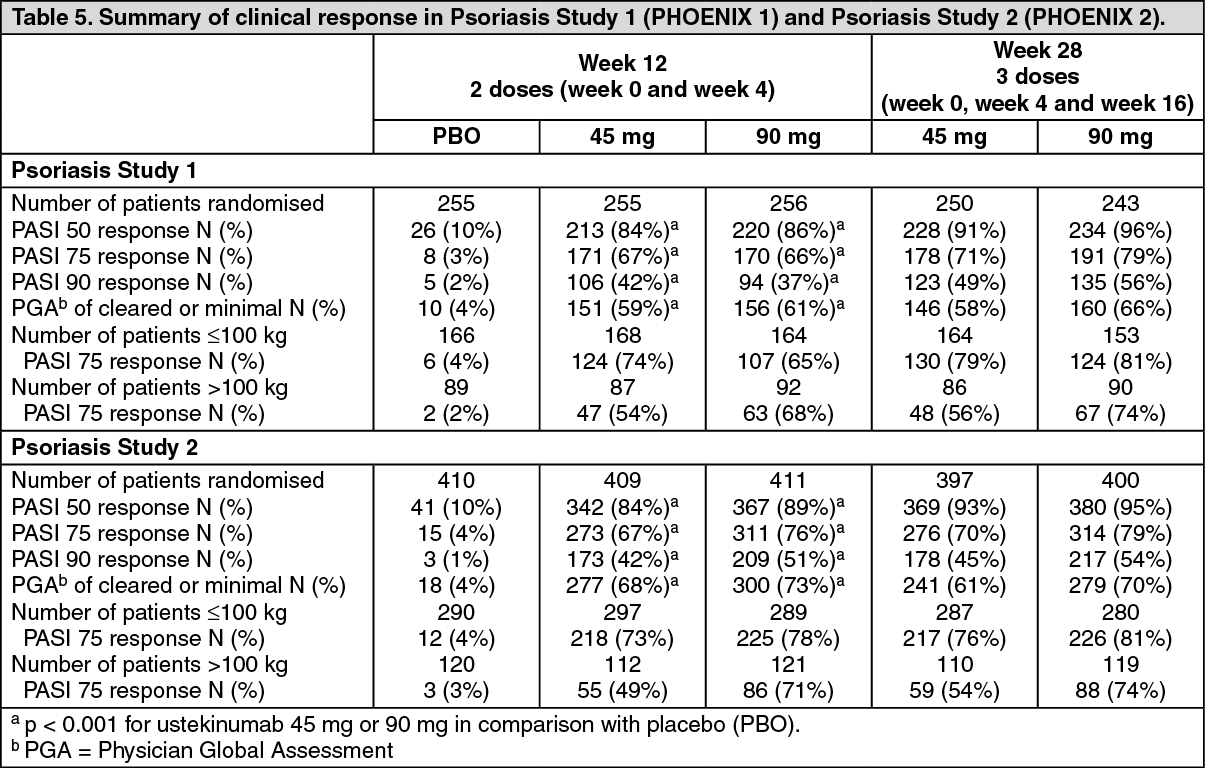 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image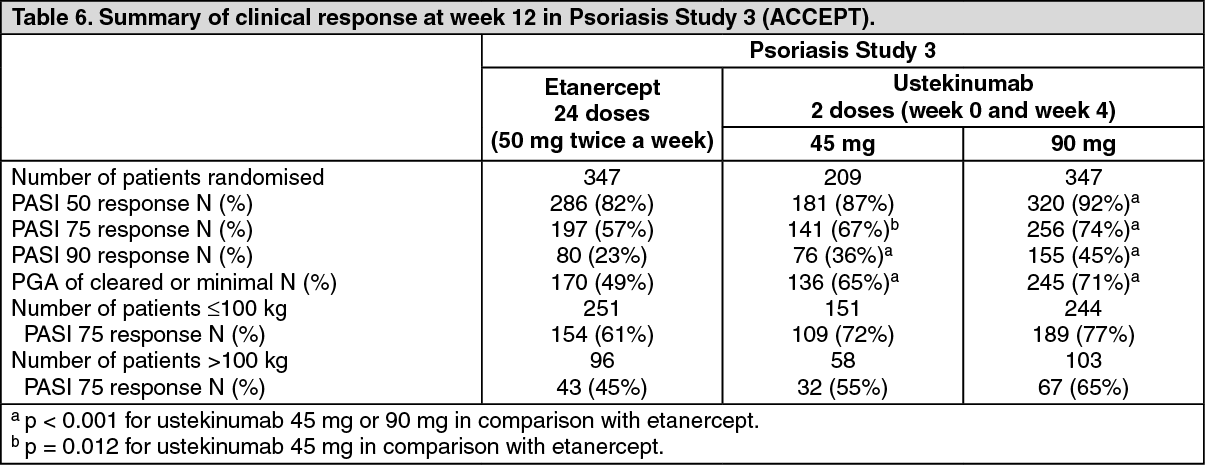 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image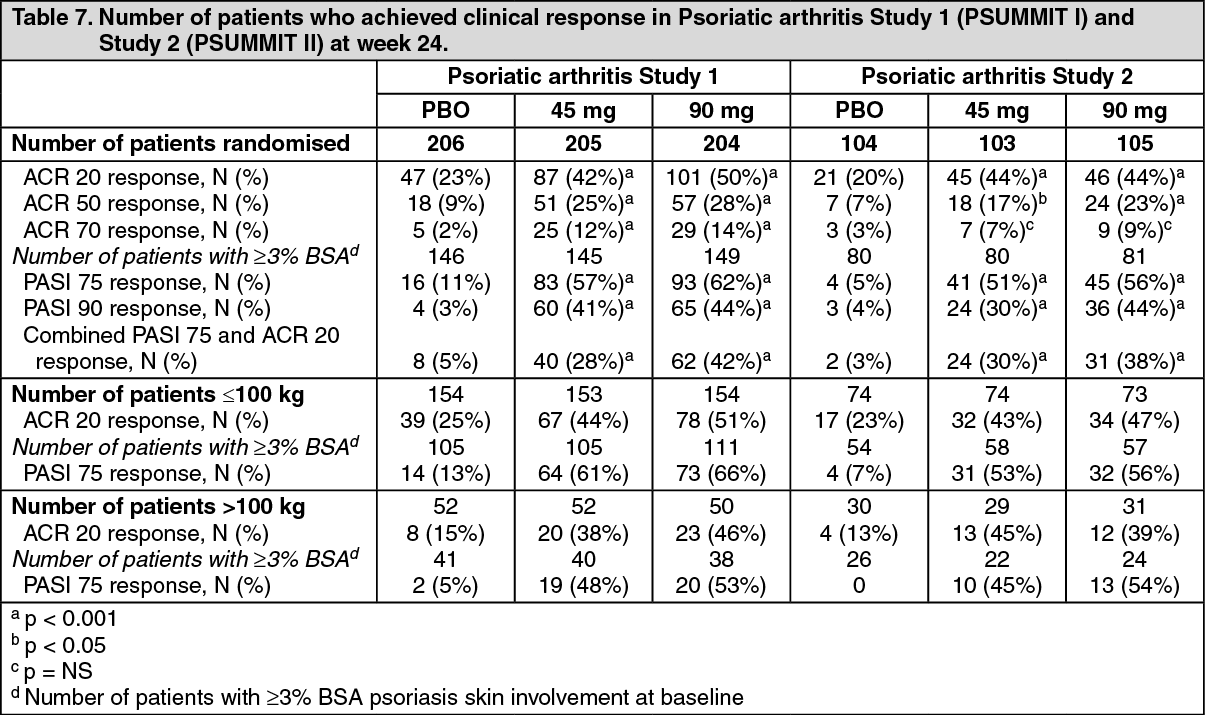 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image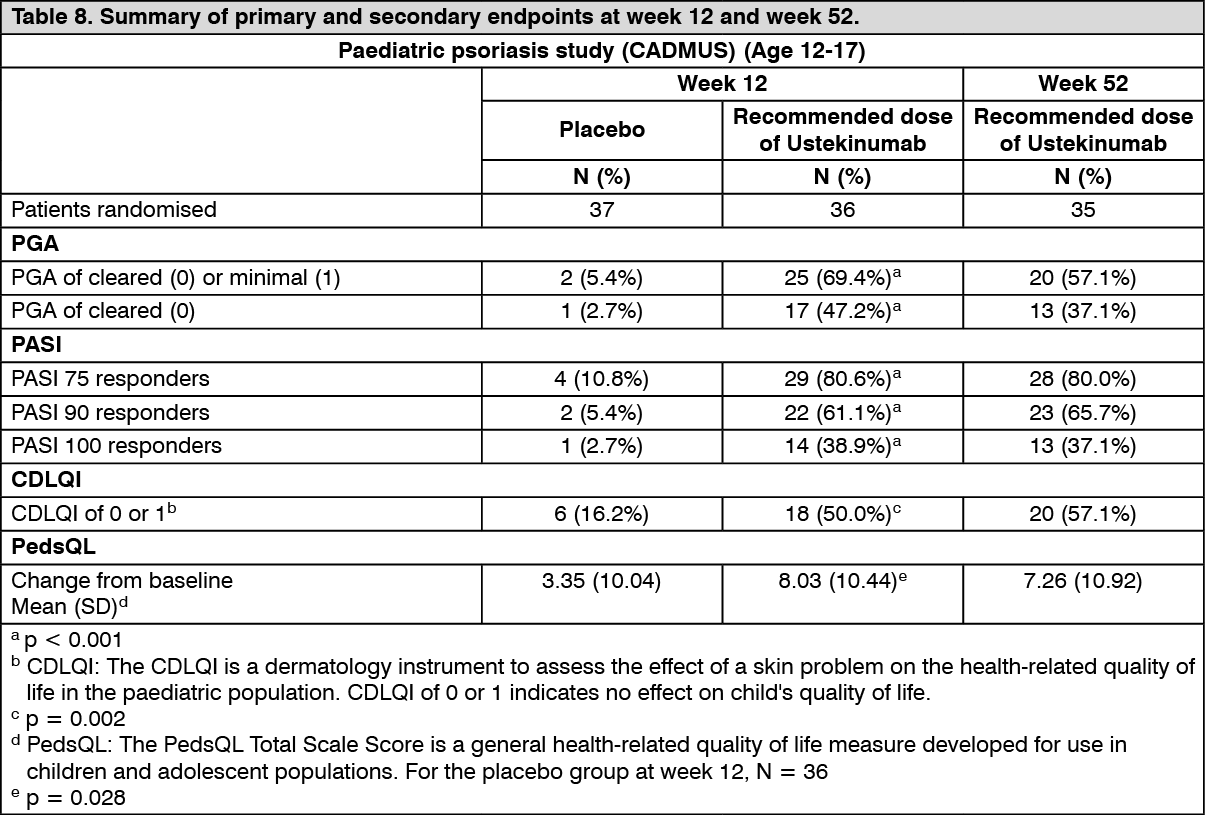 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image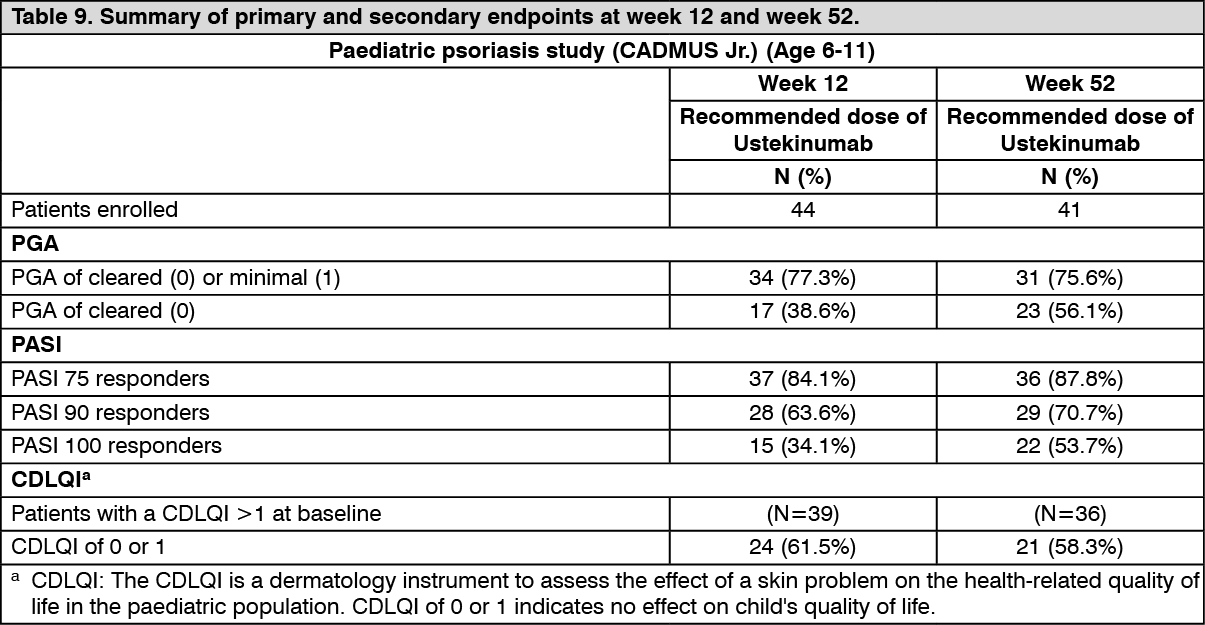 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image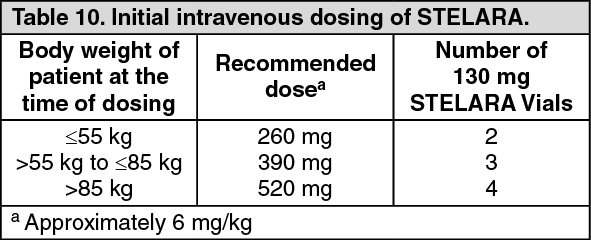 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
