


 Sign Out
Sign Out

The most frequent adverse drug reactions (ADRs) in the Rydapt arm were febrile neutropenia (83.4%), nausea (83.4%), exfoliative dermatitis (61.6%), vomiting (60.7%), headache (45.9%), petechiae (35.8%) and pyrexia (34.5%). The most frequent Grade 3/4 ADRs were febrile neutropenia (83.5%), lymphopenia (20.0%), device-related infection (15.7%), exfoliative dermatitis (13.6%), hyperglycaemia (7.0%) and nausea (5.8%). The most frequent laboratory abnormalities were haemoglobin decreased (97.3%), ANC decreased (86.7%), ALT increased (84.2%), AST increased (73.9%) and hypokalaemia (61.7%). The most frequent Grade 3/4 laboratory abnormalities were ANC decreased (85.8%), haemoglobin decreased (78.5%), ALT increased (19.4%) and hypokalaemia (13.9%).
Serious ADRs occurred at similar rates in patients in the Rydapt versus the placebo arm. The most frequent serious ADR in both arms was febrile neutropenia (16%).
Discontinuation due to any adverse reaction occurred in 3.1% of patients in the Rydapt arm versus 1.3% in the placebo arm. The most frequent Grade 3/4 adverse reaction leading to discontinuation in the Rydapt arm was exfoliative dermatitis (1.2%).
Safety profile during maintenance phase: While Table 7 provides the incidence for ADRs over the total duration of the study, when the maintenance phase (single agent Rydapt or placebo) was assessed separately, a difference in the type and severity of ADRs was observed. The overall incidence of ADRs during the maintenance phase was generally lower than during the induction and consolidation phase. Incidences of ADRs were, however, higher in the Rydapt arm than in the placebo arm during the maintenance phase. ADRs occurring more often in the midostaurin arm versus placebo during maintenance included: nausea (46.4% versus 17.9%), hyperglycaemia (20.2% versus 12.5%), vomiting (19% versus 5.4%) and QT prolongation (11.9% versus 5.4%).
Most of the haematological abnormalities reported occurred during the induction and consolidation phase when the patients received Rydapt or placebo in combination with chemotherapy. The most frequent Grade 3/4 haematological abnormalities reported in patients during the maintenance phase with Rydapt were ANC decrease (20.8% versus 18.8%) and leukopenia (7.5% versus 5.9%).
ADRs reported during the maintenance phase led to discontinuation of 1.2% of patients in the Rydapt arm and none in the placebo arm.
ASM, SM-AHN and MCL: The safety of Rydapt (100 mg twice daily) as a single agent in patients with ASM, SM-AHN and MCL was evaluated in 142 patients in two single-arm, open-label, multicentre studies. The median duration of exposure to Rydapt was 11.4 months (range: 0 to 81 months).
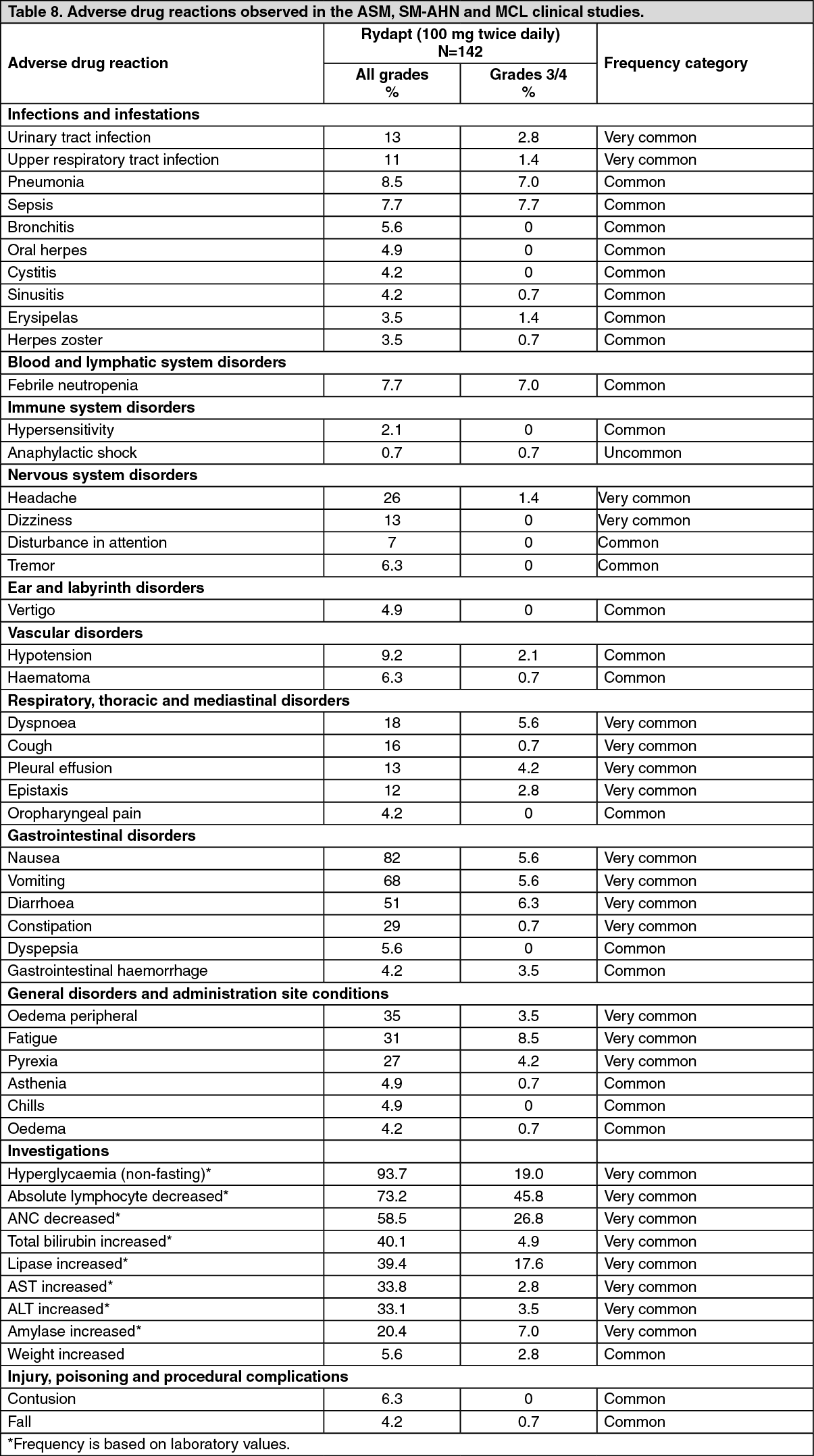
The most frequent ADRs were nausea (82%), vomiting (68%), diarrhoea (51%), peripheral oedema (35%) and fatigue (31%). The most frequent Grade 3/4 ADRs were fatigue (8.5%), sepsis (7.7%), pneumonia (7%), febrile neutropenia (7%), and diarrhoea (6.3%). The most frequent non-haematological laboratory abnormalities were hyperglycaemia (93.7%), total bilirubin increased (40.1%), lipase increased (39.4%), aspartate aminotransferase (AST) increased (33.8%), and alanine aminotransferase (ALT) increased (33.1%), while the most frequent haematological laboratory abnormalities were absolute lymphocyte count decreased (73.2%) and ANC decreased (58.5%). The most frequent Grade 3/4 laboratory abnormalities were absolute lymphocyte count decreased (45.8%), ANC decreased (26.8%), hyperglycaemia (19%), and lipase increased (17.6%).
Dose modifications (interruption or adjustment) due to ADRs occurred in 31% of patients. The most frequent ADRs that led to dose modification (incidence ≥5%) were nausea and vomiting.
ADRs that led to treatment discontinuation occurred in 9.2% of patients. The most frequent (incidence ≥1%) were febrile neutropenia, nausea, vomiting and pleural effusion.
Tabulated lists of adverse drug reactions: ADRs are listed according to MedDRA system organ class. Within each system organ class, the ADRs are ranked by frequency, with the most frequent reactions first, using the following convention (CIOMS III): very common (≥1/10); common (≥1/100 to <1/10); uncommon (≥1/1,000 to <1/100); rare (≥1/10,000 to <1/1,000); very rare (<1/10,000); not known (cannot be estimated from the available data). Within each frequency grouping, adverse reactions are presented in the order of decreasing seriousness.
AML: Table 7 presents the frequency category of ADRs reported in the phase III study in patients with newly diagnosed FLT3-mutated AML. (See Table 7.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageASM, SM-AHN and MCL: Table 8 presents the frequency category of ADRs based on pooled data from two studies in patients with ASM, SM-AHN and MCL. (See Table 8.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDescription of selected adverse drug reactions: Gastrointestinal disorders: Nausea, vomiting and diarrhoea were observed in AML, ASM, SM-AHN and MCL patients. In ASM, SM-AHN and MCL patients these events led to dose adjustment or interruption in 26% and to discontinuation in 4.2% of the patients. Most of the events occurred within the first 6 months of treatment and were managed with supportive prophylactic medicinal products.
Reporting of suspected adverse reactions: Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions.
View ADR Monitoring Form